Nome Comercial/ Apresentação
Jakavi (ruxolitinibe) 5mg / Comprimidos
Jakavi (ruxolitinibe) 10mg / Comprimidos
Jakavi (ruxolitinibe) 15mg / Comprimidos
Jakavi (ruxolitinibe) 20mg / Comprimidos
Classe Terapêutica
Agentes antineoplásicos
Indicação
Mielofibrose (MF)
É indicado para o tratamento de pacientes com mielofibrose de risco intermediário ou alto, incluindo mielofibrose primária, mielofibrose pós-policitemia vera ou mielofibrose pós trombocitemia essencial. Policitemia vera (PV)
É indicado para o tratamento de pacientes com policitemia vera que são intolerantes ou resistentes à hidroxiureia ou à terapia citorredutora de primeira linha.
Doença do enxerto contra hospedeiro (DECH) aguda
É indicado para o tratamento de pacientes com doença do enxerto contra hospedeiro aguda com 12 anos ou mais que apresentam resposta inadequada aos corticosteroides.
Doença do enxerto contra hospedeiro (DECH) crônica
É indicado para o tratamento de pacientes com doença do enxerto contra hospedeiro crônica com 12 anos ou mais que apresentam resposta inadequada aos corticosteroides ou outras terapias sistêmicas.
Dose
Para pacientes com mielofibrose (MF): A dose inicial recomendada de Jakavi para mielofibrose (MF) é baseada na contagem de plaquetas (vide tabela 5):
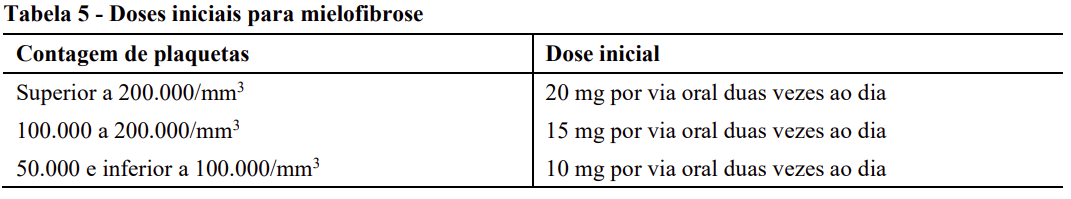 Para pacientes com policitemia vera (PV): A dose inicial recomendada de Jakavi em Policitemia vera é de 10mg administrados por via oral duas vezes por dia, dependendo da sua contagem de células sanguíneas.
Para pacientes com policitemia vera (PV): A dose inicial recomendada de Jakavi em Policitemia vera é de 10mg administrados por via oral duas vezes por dia, dependendo da sua contagem de células sanguíneas.
Para pacientes com doença do enxerto contra hospedeiro (DECH): A dose inicial recomendada em doença do enxerto contra hospedeiro é de 10mg administrados por via oral duas vezes por dia.
Modificações de dose
Para pacientes com MF: As doses podem ser tituladas com base na eficácia e segurança. Caso a eficácia seja considerada insuficiente e as contagens de células sanguíneas estiverem adequadas, as doses podem ser aumentadas em um máximo de duas doses diárias de 5mg, até a dose máxima de 25mg duas vezes ao dia. A dose inicial não deve ser aumentada nas primeiras quatro semanas de tratamento e, posteriormente, não mais frequentemente do que em intervalos de 2 semanas. O tratamento deve ser interrompido para contagens de plaquetas inferiores a 50.000/mm3 ou contagens de neutrófilos absolutos inferiores a 500/mm3. Após a recuperação das contagens sanguíneas acima destes níveis, a dosagem pode ser reiniciada a 5mg duas vezes por dia e aumentada gradualmente com base numa monitorização cuidadosa das contagens de células sanguíneas. As reduções de dose devem ser consideradas se a contagem plaquetária diminuir abaixo de 100.000/mm3 com o objetivo de evitar interrupções na dose para trombocitopenia.
Para pacientes com PV: As doses podem ser tituladas com base na eficácia e segurança. Se a eficácia é considerada insuficiente e as contagens de células sanguíneas são adequadas, as doses podem ser aumentadas num máximo de 5mg duas vezes ao dia, até a dose máxima de 25mg duas vezes ao dia. A dose inicial não deve ser aumentada nas primeiras quatro semanas de tratamento e, posteriormente, não mais frequentemente do que em intervalos de 2 semanas. O tratamento deve ser interrompido para contagens de plaquetas inferiores a 50.000/mm3 ou contagens de neutrófilos absolutos inferiores a 500/mm3. O tratamento também deve ser interrompido quando a hemoglobina estiver abaixo de 8g/dL. Após a recuperação das contagens sanguíneas acima destes níveis, a dosagem pode ser reiniciada a 5mg duas vezes por dia e aumentada gradualmente com base numa monitorização cuidadosa das contagens de células sanguíneas. A redução da dose deve ser considerada se a contagem das plaquetas diminuírem durante o tratamento, conforme destacado na tabela 6, com o objetivo de evitar interrupções de dose para trombocitopenia:
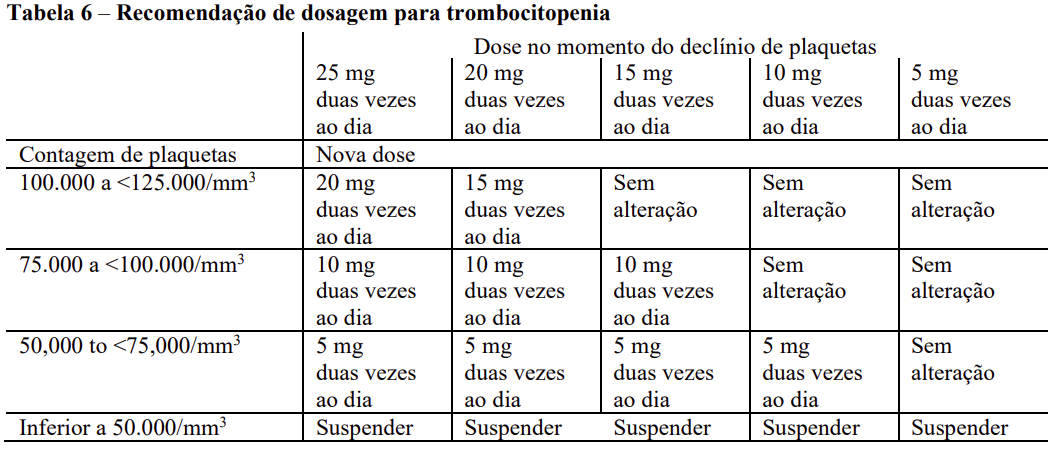
Na policitemia vera, a redução da dose também deve ser considerada se a hemoglobina diminuir abaixo de 12g/dL e é recomendada se a hemoglobina diminuir abaixo de 10g/dL.
Para pacientes com DECH: Podem ser necessárias reduções de dose e interrupções temporárias do tratamento em pacientes com DECH com trombocitopenia, neutropenia ou bilirrubina total elevada após terapia de suporte padrão incluindo fatores de crescimento, terapias anti-infecciosas e transfusões. Recomenda-se etapa de redução de um nível de dose (10mg duas vezes por dia para 5mg duas vezes por dia ou 5mg duas vezes por dia para 5mg uma vez por dia). Em pacientes que são incapazes de tolerar com uma dose de 5mg uma vez ao dia, o tratamento deve ser interrompido.
As recomendações detalhadas de dosagem são fornecidas na Tabela 7.
Tabela 7 – Recomendações de dosagem para pacientes com trombocitopenia, neutropenia ou bilirrubina total elevada em pacientes com doença do enxerto contra hospedeiro
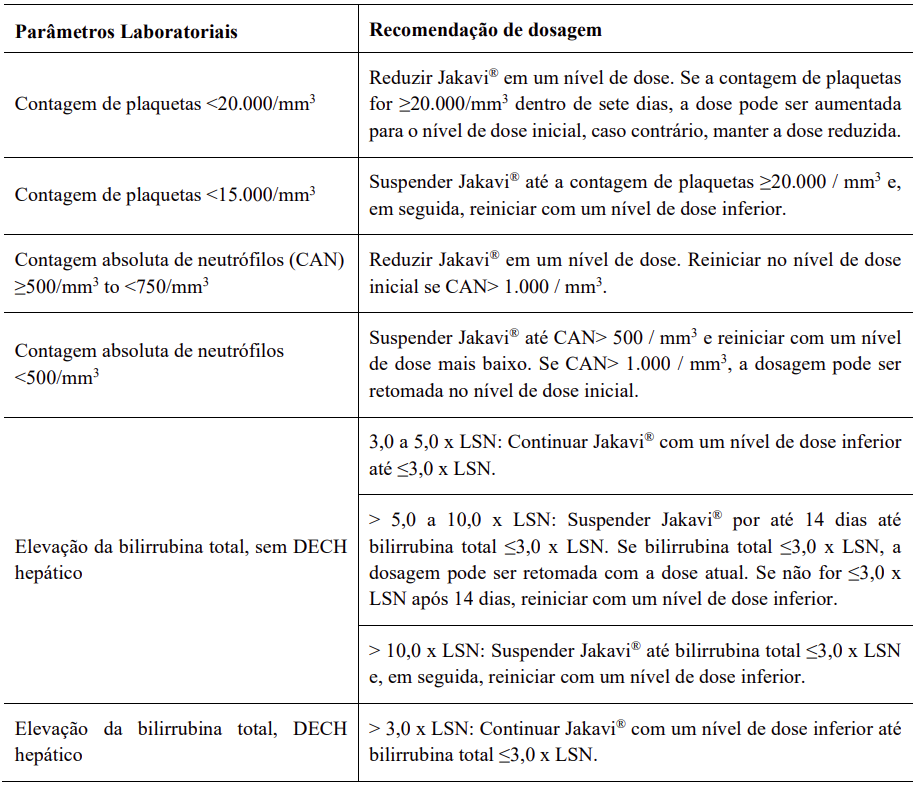
Ajuste da dose em caso de uso concomitante com inibidores potentes da CYP3A4 ou fluconazol – pacientes com MF, PV e DECH: Quando é administrado com inibidores potentes da CYP3A4 em pacientes com MF e PV ou inibidores duplos moderados das enzimas CYP2C9 e CYP3A4 (ex. fluconazol) em pacientes com MF, PV e DECH, a dose diária total deve ser reduzida em aproximadamente 50% tanto pela redução na dose de duas vezes ao dia, ou reduzindo a frequência da dose para uma dose diária única correspondente quando a dosagem diária de duas vezes ao dia não for possível. O uso concomitante com fluconazol em doses superiores a 200mg por dia devem ser evitados. O monitoramento mais frequente dos parâmetros hematológicos e sinais e sintomas clínicos relacionados a reações adversas ao medicamento (RAMs) é recomendado com o início de um potente inibidor da CYP3A4 ou um inibidor duplo moderado das enzimas CYP2C9 e CYP3A4. Se a contagem de plaquetas diminuir para menos de 100.000/mm3, o uso concomitante deve ser evitado.
Ajuste de dose
– Insuficiência renal – pacientes com MF
Em pacientes com insuficiência renal grave (clearance de creatinina (Clcr) menor que 30mL/min), a dose inicial recomendada baseada nas contagens de plaquetas para pacientes com mielofibrose (MF) deve ser reduzida em aproximadamente 50% a ser administrada duas vezes ao dia. Há dados limitados para determinar as melhores opções de dosagem para pacientes com doença renal em estágio final (ESRD) em diálise. Os dados disponíveis nesta população sugerem que pacientes com MF em diálise devem iniciar com uma dose única de 15 ou 20mg baseada nas contagens de plaquetas com doses únicas subsequentes somente depois de cada sessão de diálise e com o monitoramento cuidadoso da segurança e da eficácia.
– Insuficiência renal – pacientes com PV e DECH
A dose inicial recomendada para pacientes com policitemia vera (PV) e doença do enxerto contra hospedeiro (DECH) com insuficiência renal grave é de 5mg duas vezes ao dia. Os pacientes diagnosticados com insuficiência renal grave (Clcr menor que 30mL/min) devem ser cuidadosamente monitorados e podem precisar ter suas doses reduzidas para evitar RAMs. Há dados limitados para determinar as melhores opções de dosagem para pacientes com doença renal em estágio final (ESRD) em diálise. A dose inicial recomendada para pacientes com PV e DECH com ESRD em hemodiálise é uma dose única de 10g (ou duas doses de 5 mg a cada 12 horas), a ser administrada pós-diálise e apenas no dia de hemodiálise. Estas recomendações de dose baseiam-se em simulações e qualquer modificação de dose em pacientes com ESRD deve ser seguida com uma monitoração cuidadosa da segurança e eficácia. Não existem dados disponíveis para pacientes submetidos à diálise peritoneal ou hemofiltração venovenosa contínua.
Insuficiência hepática – pacientes com MF, PV e DECH
Em pacientes com MF com qualquer comprometimento hepático, a dose inicial recomendada baseada na contagem de plaquetas deve ser reduzida em aproximadamente 50% a ser administrada duas vezes ao dia. A dose inicial recomendada para pacientes com PV é de 5mg duas vezes ao dia. As doses subsequentes devem ser ajustadas com base em um monitoramento cuidadoso da segurança e da eficácia. Os pacientes diagnosticados com insuficiência hepática, enquanto receberem Jakavi®, devem ser cuidadosamente monitorados e podem precisar diminuir a dose para evitar RAMs. Devem ser realizados hemogramas completos, incluindo a contagem diferencial de leucócitos, monitorados pelo menos a cada uma a duas semanas durante as primeiras 6 semanas após o início do tratamento, e conforme indicado clinicamente após a sua função hepática e as contagens sanguíneas se estabilizarem. A dose pode ser titulada para reduzir o risco de citopenia. Em pacientes com DECH com qualquer comprometimento hepático, incluindo DECH hepático, nenhuma modificação da dose inicial é recomendada. Em pacientes com DECH com envolvimento hepático e aumento da bilirrubina total para > 3xLSN, as contagens sanguíneas devem ser monitoradas com mais frequência para toxicidade e uma redução de um nível de dose pode ser considerada.
Vias de administração
ORAL
Administração
É administrado oralmente e pode ser administrado com ou sem alimento.
Cuidados específicos e monitoramento
Gravidez, lactação e mulheres e homens com potencial reprodutivo
– Gravidez
Não existem estudos adequados e bem controlados em mulheres grávidas. Estudos de reprodução em ratos e coelhos demonstraram que o ruxolitinibe induz embriotoxicidade e fetotoxicidade. Seguindo a exposição pré-natal, há aumento na perda pós-implantação em coelhos e redução do peso fetal em ratos e coelhos foi observado. Em ratos e coelhos esses efeitos ocorreram em exposições de aproximadamente 2 vezes e 0,07 vezes, respectivamente em relação as condições clínicas na dose máxima recomendada a humanos de 25mg duas vezes ao dia com base na AUC. O uso durante a gravidez não é recomendado. A paciente deve ser aconselhada sobre os riscos para o feto caso Jakavi® seja usado durante a gravidez ou se a paciente engravidar durante o tratamento.
– Lactação
Não se sabe se o ruxolitinibe é transferido para o leite humano. Não há dados sobre os efeitos do ruxolitinibe na criança amamentada ou os efeitos do ruxolitinibe na produção de leite. O ruxolitinibe e/ou seus metabólitos passaram rapidamente para o leite de ratas lactantes. Devido ao potencial de reações adversas ao medicamento graves em lactentes, deve-se tomar uma decisão sobre interromper a amamentação ou descontinuar o medicamento, levando em consideração a importância do medicamento para a mãe. Recomenda-se que as mulheres não amamentem durante o tratamento. Uso criterioso no aleitamento ou na doação de leite humano.
– Contracepção
Mulheres com potencial para engravidar devem ser informadas que estudos em animais foram realizados demonstrando que o ruxolitinibe pode ser prejudicial para o desenvolvimento do feto. Mulheres com potencial para engravidar sexualmente ativas devem utilizar métodos contraceptivos efetivos durante o tratamento. Em caso de gravidez, devem ser realizadas avaliações de risco/benefício individuais, com aconselhamento cuidadoso em relação ao risco potencial para o feto com base nos dados mais recentes disponíveis.
-Infertilidade
Estudos em animais, demonstraram que, nenhum efeito foi observado na fertilidade ou no potencial para reprodução em ratos machos e fêmeas. Em um estudo pré e pós-natal em ratos, a fertilidade na primeira ninhada também não foi afetada.
Efeitos na habilidade de dirigir veículos e/ou operar máquinas
Não tem efeito sedativo, ou é insignificante. Entretanto, pacientes que apresentaram tonturas durante o tratamento não devem conduzir veículos ou operar máquinas, pois sua habilidade e atenção podem estar prejudicadas.
Atenção: Contém lactose
Interações medicamentosas
Agentes que podem alterar a concentração plasmática de ruxolitinibe
– Inibidores potentes da CYP3A4 (tais como boceprevir, telaprevir, claritromicina, itraconazol, cetoconazol, posaconazol, indinavir, lopinavir/ritonavir, ritonavir, nefazodona, nelfinavir, saquinavir, telitromicina, voriconazol, mas não limitado a esses): em indivíduos sadios recebendo cetoconazol, um potente inibidor da CYP3A4, com 200mg duas vezes ao dia por quatro dias, a AUC de ruxolitinibe aumentou em 91% e a meia vida foi prolongada de 3,7 para 6,0 horas. Ao administrar com inibidores potentes da CYP3A4, a dose diária total deve ser reduzida em aproximadamente 50%, exceto em pacientes com DECH. O efeito de inibidores potentes da CYP3A4 em pacientes com DECH não teve um impacto significativo em qualquer parâmetro no modelo farmacocinético da população. Os pacientes devem ser cuidadosamente monitorados quanto a citopenias e a dose titulada com base na segurança e na eficácia.
– Inibidores leves ou moderados da CYP3A4 (tais como ciprofloxacina, eritromicina, atazanavir, diltiazem, cimetidina, mas não limitado a esses): em indivíduos sadios recebendo eritromicina, um inibidor moderado da CYP3A4, a 500mg duas vezes ao dia por quatro dias, houve um aumento de 27% na AUC de ruxolitinibe. Não é recomendado nenhum ajuste de dose quando é coadministrado com inibidores leves ou moderados da CYP3A4 (ex.: eritromicina). Os pacientes devem ser cuidadosamente monitorados quanto a citopenias ao iniciar uma terapia com um inibidor moderado da CYP3A4.
– Inibidores duplos moderados da CYP2C9 e CYP3A4 (ex. fluconazol): em indivíduos sadios recebendo fluconazol, um inibidor duplo de CYP2C9 e CYP3A4, em uma única dose de 400mg seguida de uma dose de 200mg uma vez ao dia por sete dias, houve um aumento de 232% na AUC de ruxolitinibe. Uma redução da dose de 50% deve ser considerada quando utilizar medicamentos que são inibidores duplos das enzimas CYP2C9 e CYP3A4. O uso concomitante com doses de fluconazol superiores a 200mg por dia deve ser evitado.
– Indutores da CYP3A4 (tais como carbamazepina, fenobarbital e outros antiepilépticos, fenitoína, rifampicina, erva de São João (Hypericum perforatum), mas não limitado a esses): após início de um indutor da CYP3A4, nenhum ajuste de dose é recomendado. Aumentos graduais na dose podem ser considerados se a eficácia da terapia diminuir durante o tratamento com um indutor da CYP3A4. Em indivíduos sadios recebendo rifampicina, um potente indutor da CYP3A4, a 600mg uma vez ao dia por 10 dias, a AUC de ruxolitinibe após uma única dose diminuiu em 71% e a meia-vida diminuiu de 3,3 para 1,7 horas. A quantidade relativa de metabólitos ativos aumentou em relação ao composto original.
– Glicoproteína p e outros transportadores: pode inibir a glicoproteína-p e a proteína BCRP (Breast Cancer Resistance Protein) no intestino. Isto pode resultar em um aumento da exposição sistêmica de substratos destes transportadores, tais como dabigatrano etexilato, ciclosporina, rosuvastatina e, potencialmente, digoxina. Recomenda-se monitoramento terapêutico do fármaco ou monitoramento clínico da substância afetada. É possível que a potencial inibição da gp-p e da BCRP no intestino possa ser minimizada se o intervalo de tempo entre administrações for o mais longo possível.
Interações estudadas com outros medicamentos
– Substratos da CYP3A4: Um estudo em indivíduos sadios indicou que não apresentou significativa interação farmacocinética com midazolam (substrato da CYP3A4).
– Contraceptivos orais: Um estudo com indivíduos sadios indicou que não afeta a farmacocinética de contraceptivos orais contendo etinilestradiol e levonorgestrel. Desta forma, não se pode prever que a eficácia contraceptiva desta combinação será comprometida pela coadministração de ruxolitinibe.
Fatores de crescimento hematopoiético e terapias citorredutoras
O uso concomitante de terapias citorredutoras ou fatores de crescimento hematopoiético não foi estudado. A segurança e eficácia destas coadministrações são desconhecidas.
Estabilidade/ Conservação
Conservar em temperatura ambiente (entre 15 e 30°C).
Reações adversas
Resumo tabulado de reações adversas ao medicamento provenientes de estudos clínicos
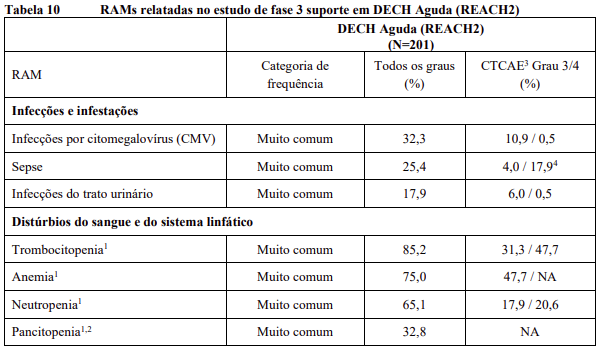
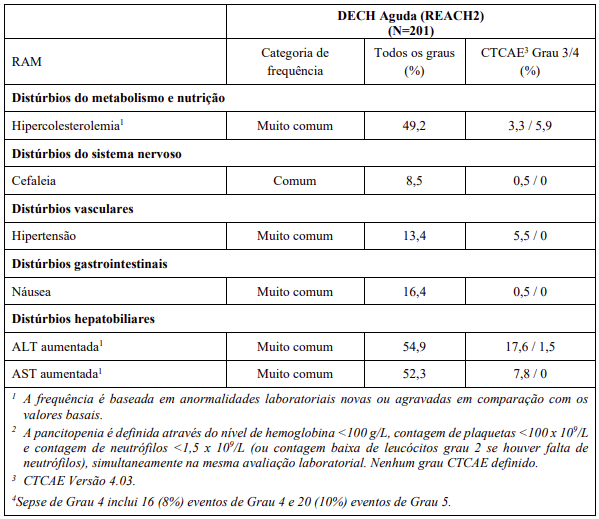
As RAMs a partir de estudos clínicos em MF e PV são listadas na Tabela 8. As RAMs a partir de estudos clínicos em DECH aguda e crônica estão listadas na Tabela 9 e as RAMs do estudo suporte na Tabela 10. Todas as RAMs estão listadas de acordo com a classe de sistema de órgão (SOC) do MedDRA.
Dentro de cada classe de sistema de órgão, as RAMs são classificadas pela frequência, com as reações adversas a medicamentos mais frequentes aparecendo primeiro. Além disso, a categoria de frequência correspondente para cada RAM baseia-se na convenção a seguir (CIOMS III): muito comum (≥ 1/10); comum (≥ 1/100 a < 1/10); incomum (≥ 1/1.000 a < 1/100); rara (≥ 1/10.000 a < 1/1.000); muito rara (< 1/10.000). No programa de estudos clínicos a gravidade das RAMs foi avaliada com base nos CTCAE que definem como Grau 1 = leve, Grau 2 = moderada, Grau 3 = Grave e Grau 4 = ameaça à vida ou incapacitante, Grau 5 = morte.
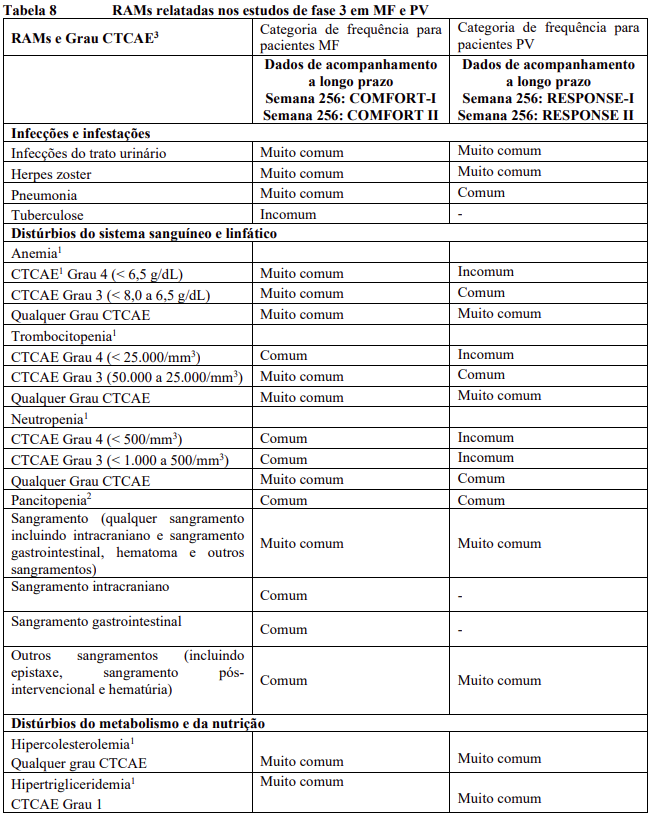
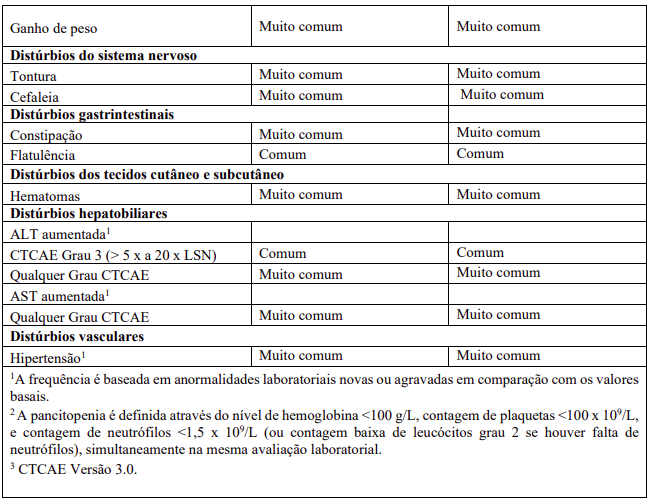
Após descontinuação, os pacientes com MF podem apresentar um retorno dos sintomas de mielofibrose, tais como fadiga, dor óssea, febre, prurido, sudorese noturna, esplenomegalia sintomática e perda de peso. Em estudos clínicos de MF, a pontuação total de sintomas para mielofibrose retornaram gradualmente para os valores do basal em até 7 dias após a descontinuação da dose.
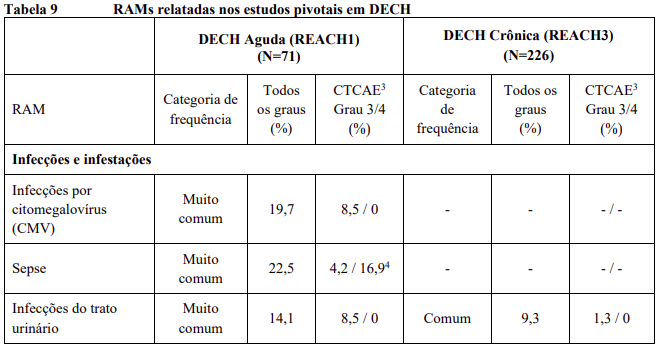
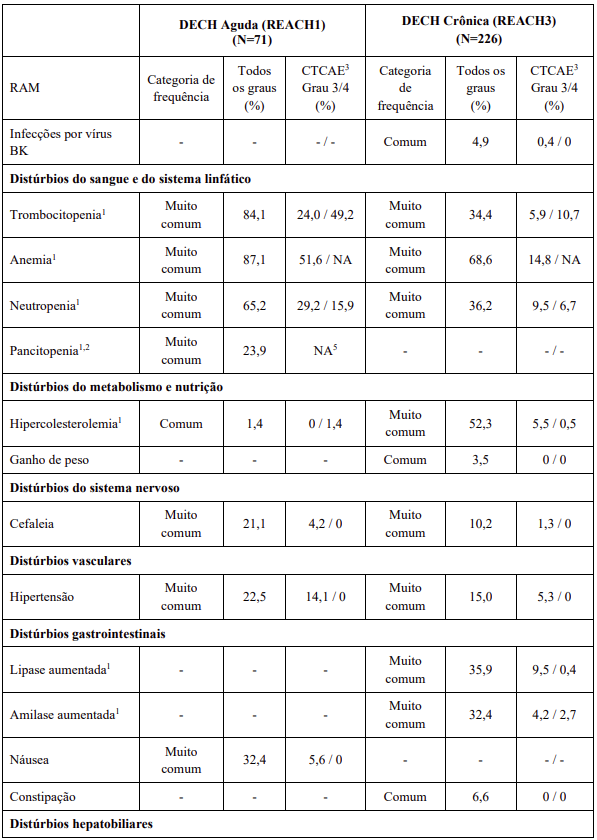
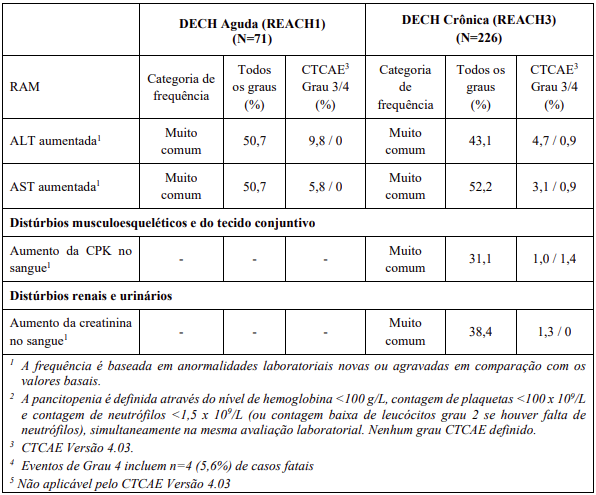
RAMs a partir de relatos espontâneos e casos de literatura (frequência desconhecida)
A tuberculose como RAM tem sido observada na pós-comercialização em pacientes com PV através de relatos de casos espontâneos e da literatura. Como essas reações são relatadas voluntariamente por uma população de tamanho incerto, nem sempre é possível estimar com segurança sua frequência, que é portanto, caracterizada como desconhecida.
Contraindicações
Hipersensibilidade ao princípio ativo ou a algum dos excipientes.
Fonte:
Jakavi®. [Bula]. São Paulo: Novartis Biociências S.A. Disponível em: https://consultas.anvisa.gov.br/#/bulario/q/?numeroRegistro=100681121: 28/08/2025