Nome Comercial/ Apresentação
Tasigna (nilotinibe) 150mg / Cápsulas
Classe Terapêutica
Agentes antineoplásicos – inibidor da proteína tirosinoquinase
Indicação
Tasigna® é indicado para:
- o tratamento de pacientes adultos com leucemia mieloide crônica cromossomo Philadelphia positivo (LMC Ph+) em fase crônica (FC) recém-diagnosticada.
- o tratamento de pacientes adultos com leucemia mieloide crônica cromossomo Philadelphia positivo (LMC Ph+) em fase crônica (FC) ou em fase acelerada após falha ou intolerância a pelo menos uma terapia prévia, incluindo imatinibe.
Dose
População alvo geral
Posologia em pacientes com LMC Ph+-FC recém-diagnosticada
A dose recomendada de Tasigna® é de 300 mg duas vezes ao dia. O tratamento deverá prosseguir enquanto o paciente continuar a ser beneficiado.
Posologia em pacientes com LMC Ph+-FC recém-diagnosticada que tenham atingido resposta molecular profunda sustentada (RM 4,5 log)
Elegibilidade para descontinuação do tratamento
A descontinuação do tratamento pode ser considerada em pacientes com LMC Ph+-FC elegíveis, que tenham sido tratados com Tasigna® por, no mínimo, 3 anos e se a resposta molecular profunda (RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI)) for sustentada por, no mínimo, um ano, imediatamente antes da descontinuação da terapia. A descontinuação de Tasigna® deve ser iniciada por um médico experiente no tratamento de pacientes com LMC.
Considerar a descontinuação do tratamento em pacientes com LMC Ph+-FC recém-diagnosticada quando:
• tenham sido tratados com Tasigna® por, no mínimo, 3 anos;
• tenham resposta molecular RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI) sustentada por, no mínimo, um ano, imediatamente antes da descontinuação da terapia;
• tenham atingido RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI) na última avaliação realizada imediatamente antes da descontinuação do tratamento;
• existir a confirmação de que há a expressão da transcrição BCR-ABL típica (e13a2/b2a2 ou e14a2/b3a2);
• não houver histórico de fase acelerada ou crise blástica;
• não houver histórico de tentativas anteriores de descontinuação devido a remissão livre de tratamento que resultaram em recaída.
A recaída molecular pode ocorrer durante a fase livre de tratamento, e dados de resultados a longo prazo ainda não estão disponíveis. Pacientes elegíveis a descontinuar a terapia com Tasigna® devem ser monitorados quanto aos níveis de transcrição BCR-ABL e a contagem sanguínea completa mensalmente, por um ano, e, posteriormente, a cada 6 semanas no segundo ano e a cada 12 semanas do terceiro ano em diante. O monitoramento dos níveis de transcrição BCR-ABL deve ser realizado com um teste de diagnóstico quantitativo validado para medir os níveis de resposta molecular na Escala Internacional (EI) com uma sensibilidade de pelo menos RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI). Para pacientes que perdem RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI), mas não resposta molecular maior (RMM = (BCR-ABL/ABL ≤ 0,1% EI)) durante a fase livre de tratamento, os níveis de transcrição BCR-ABL devem ser monitorados a cada 2 semanas até que os níveis BCR-ABL retornem a um intervalo entre RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI) e RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI). Os pacientes que mantêm os níveis de BCR-ABL entre RMM (BCR-ABL/ABL ≤ 0,1% EI) e RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI) por um mínimo de 4 medições consecutivas podem retornar ao cronograma de monitoramento original.
Reinício do tratamento em pacientes que perderam Resposta Molecular Profunda após descontinuação da terapia com Tasigna®
Pacientes que apresentarem perda de RMM (BCR-ABL/ABL ≤ 0,1% EI) devem reiniciar o tratamento dentro de 4 semanas a partir da verificação da ocorrência da perda de remissão. A terapia com Tasigna® deve ser reiniciada na dose de 300 mg duas vezes ao dia ou em dose reduzida de 400 mg uma vez ao dia caso tenha havido redução de dose anterior à descontinuação da terapia. Pacientes que reiniciaram a terapia com Tasigna® devem monitorar os níveis de transcrição BCR-ABL mensalmente até que a RMM (BCR-ABL/ABL ≤ 0,1% EI) seja reestabelecida.
Posologia em pacientes com LMC Ph+-FC e LMC Ph+-FA) após resistência ou intolerância a pelo menos uma terapia prévia, incluindo imatinibe
A dose recomendada de Tasigna® é de 400 mg duas vezes ao dia. O tratamento deverá prosseguir enquanto o paciente continuar a ser beneficiado.
Posologia em pacientes LMC Ph+-FC tratados com Tasigna®, após terapia prévia com imatinibe, que tenham atingido resposta molecular profunda sustentada (RM 4,5 log)
Elegibilidade para descontinuação do tratamento
A descontinuação de tratamento pode ser considerada em pacientes com LMC Ph+-FC que tenham sido tratados com Tasigna® por, no mínimo, 3 anos e se a resposta molecular profunda (RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI)) for sustentada por, no mínimo, um ano, imediatamente antes da descontinuação da terapia. A descontinuação de Tasigna® deve ser iniciada por um médico experiente no tratamento de pacientes com LMC.
Considerar descontinuação do tratamento em pacientes com LMC Ph+-FC que tenham sido tratados com imatinibe ou que sejam resistentes ou intolerantes ao tratamento com imatinibe, e que tenham atingido resposta molecular profunda (RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI)) com Tasigna® quando:
• tenham sido tratados com Tasigna® por, no mínimo, 3 anos;
• tenham atingido resposta molecular RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI);
• tenham atingido resposta molecular sustentada RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI) por, pelo menos, um ano antes da descontinuação do tratamento;
• existir a confirmação de que há a expressão da transcrição BCR-ABL típica (e13a2/b2a2 or e14a2/b3a2);
• não houver histórico de fase acelerada ou crise blástica;
• não houver histórico de tentativas anteriores de descontinuação devido a remissão livre de tratamento que resultaram em recaída.
A recaída molecular pode ocorrer durante a fase livre de tratamento, e dados de resultados a longo prazo ainda não estão disponíveis.
Pacientes elegíveis a descontinuar a terapia com Tasigna® devem ser monitorados quanto aos níveis de transcrição BCR-ABL e a contagem sanguínea completa mensalmente, por um ano, e, posteriormente, a cada 6 semanas no segundo ano e a cada 12 semanas do terceiro ano em diante. O monitoramento dos níveis de transcrição BCR-ABL deve ser realizado com um teste de diagnóstico quantitativo validado para medir os níveis de resposta molecular na Escala Internacional (EI) com uma sensibilidade de pelo menos RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI).
Reinício do tratamento em pacientes que perderam Resposta Molecular Profunda após descontinuação da terapia com Tasigna®
Pacientes com perda de RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI) confirmada (duas medidas consecutivas separadas por pelo menos 4 semanas apresentando perda de RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI)) ou perda de RMM (BCRABL/ABL ≤ 0,1% EI) devem reiniciar o tratamento dentro de 4 semanas a partir da verificação da ocorrência da perda de remissão. A terapia com Tasigna® deve ser reiniciada na dose de 300 mg ou 400 mg duas vezes ao dia. Pacientes que reiniciaram a terapia com Tasigna® devem ser monitorados quanto aos níveis de transcrição BCR-ABL mensalmente até a RMM (BCR-ABL/ABL ≤ 0,1% EI) prévia ou RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI) ser reestabelecida.
Ajuste de dose
O eletrocardiograma (ECG) basal é recomendado antes do início da terapia com Tasigna® e deve ser repetido após 7 dias e conforme clinicamente indicado. Hipocalemia ou hipomagnesemia devem ser corrigidas antes da administração de Tasigna® e os níveis sanguíneos de potássio e magnésio devem ser monitorados periodicamente durante a terapia, particularmente em pacientes em risco para anormalidades eletrolíticas. Foram relatados com o tratamento com Tasigna® aumento dos níveis séricos de colesterol total. O perfil lipídico deve ser determinado antes de iniciar o tratamento com Tasigna® e, avaliado aos 3 e 6 meses após o início do tratamento, e pelo menos anualmente durante o tratamento crônico. Foram relatados aumento dos níveis de glicose sanguínea na terapia com Tasigna®. Os níveis de glicose sanguínea devem ser avaliados antes do início do tratamento com Tasigna® e monitorados durante o tratamento. Devido à possível ocorrência de Síndrome de Lise Tumoral (SLT) são recomendadas, correção de desidratação clinicamente significante e tratamento de níveis elevados de ácido úrico, antes do início da terapia com Tasigna®. Tasigna® pode ser interrompido temporariamente e/ou sua dose reduzida em decorrência de toxicidades hematológicas (neutropenia, trombocitopenia) que não estejam relacionadas à leucemia subjacente (vide Tabela12).

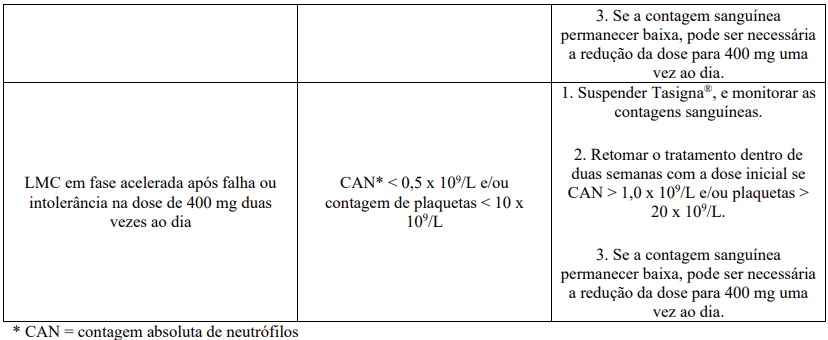
Se ocorrer toxicidade não hematológica, moderada ou grave, clinicamente significante, o tratamento deve ser interrompido e poderá ser retomado com a dose de 400mg uma vez ao dia, desde que a toxicidade esteja resolvida. Se clinicamente apropriado, o re-escalonamento da dose para 300mg (LMC Ph+-FC recém-diagnosticado) ou 400 mg (LMC Ph+-FC e LMC-FA após falha ou intolerância) duas vezes ao dia deve ser considerado.
Lipase sérica elevada: para elevações de lipase de Graus 3 e 4, as doses devem ser reduzidas para 400 mg uma vez ao dia ou interrompidas. Os níveis de lipase sérica devem ser testados mensalmente, ou quando clinicamente indicado.
Bilirrubina e transaminases hepáticas elevadas: para elevações de bilirrubina ou transaminases hepáticas de Graus 3 e 4, as doses devem ser reduzidas para 400 mg uma vez ao dia ou interrompidas. Os níveis de bilirrubina e de transaminases hepáticas devem ser testados mensalmente ou quando clinicamente indicado.
População especial
Pacientes pediátricos (abaixo de 18 anos)
Dados de segurança e eficácia em crianças e adolescentes abaixo de 18 anos de idade não foram estabelecidos.
Pacientes geriátricos (≥ 65 anos de idade ou mais)
Aproximadamente 12% e 30% dos pacientes nos estudos clínicos (LMC Ph+-FC recém-diagnosticada e LMC Ph+-FC e LMC-FA após falha ou intolerância) tinham 65 anos de idade ou mais. Não foram observadas grandes diferenças na segurança e eficácia em pacientes com idade ≥ 65 anos quando comparados com pacientes adultos de 18 a 65 anos de idade.
Insuficiência renal
Não foram realizados estudos clínicos envolvendo pacientes com insuficiência na função renal. Os estudos clínicos excluíram pacientes com concentração de creatinina sérica > 1,5 vezes o limite superior da normalidade. Como o nilotinibe e seus metabólitos não são excretados por via renal, não é esperada uma diminuição no clearance (depuração) corpóreo total em pacientes com insuficiência renal.
Insuficiência hepática
A insuficiência hepática tem um efeito modesto na farmacocinética do nilotinibe. O ajuste de dose não é considerado necessário em pacientes com insuficiência hepática, mas estes devem ser tratados com cautela.
Distúrbios cardíacos
Nos estudos clínicos foram excluídos pacientes com doenças cardíacas não controladas ou significantes, incluindo infarto recente do miocárdio, insuficiência cardíaca congestiva, angina instável, ou bradicardia clinicamente significantes.
Deve-se ter cautela com pacientes com distúrbios cardíacos relevantes.
Vias de administração
ORAL
Administração
Este medicamento não deve ser partido ou mastigado.
Tasigna® deve ser tomado duas vezes ao dia, em intervalos de 12 horas, aproximadamente, e não deve ser ingerido com alimentos. As cápsulas devem ser ingeridas inteiras com água. Nenhum alimento deve ser consumido por, pelo menos, duas horas antes da dose ser ingerida e nenhum alimento deve ser consumido por, pelo menos, uma hora após a ingestão da dose.
Para pacientes com dificuldade de deglutição, o conteúdo de cada cápsula deve ser disperso em uma colher de chá de suco de maçã e deve ser ingerido imediatamente. Não mais do que uma colher de chá de suco de maçã e nenhum outro alimento deve ser usado.
Se o paciente esquecer de tomar uma dose, ele não deve tomar uma dose adicional, mas deve tomar a próxima dose como de costume.
Cuidados específicos e monitoramento
Mielossupressão
O tratamento com Tasigna® está frequentemente associado à trombocitopenia, neutropenia e anemia (NCI CTC – National Cancer Institute Common Toxicity Criteria – Graus 3/4). Esta ocorrência é mais frequente em pacientes com LMC após falha ou intolerância ao imatinibe e em particular em pacientes com LMC-FA. Contagem sanguínea completa deve ser realizada a cada duas semanas nos dois primeiros meses e depois, mensalmente, ou quando clinicamente indicado. Mielossupressão foi geralmente reversível e normalmente controlada com interrupção temporária do tratamento com Tasigna® ou diminuição da dose.
Prolongamento do intervalo QT
Dados in vitro sugerem que nilotinibe tem o potencial de prolongar a repolarização ventricular cardíaca (intervalo QT).
No estudo de fase III em pacientes com LMC Ph+-FC recém-diagnosticada, a mudança observada no tempo médio do intervalo QTcF, a partir do nível basal, no estado de equilíbrio do grupo de nilotinibe 300 mg duas vezes ao dia foi de 6 ms. Na dose recomendada de 300 mg duas vezes ao dia, nenhum paciente teve a QTcF absoluta > 480 ms e nenhum evento de Torsade de Pointes foi observado.No estudo de fase II com pacientes com LMC após falha ou intolerância ao imatinibe em fase crônica e acelerada, tratados com nilotinibe 400 mg duas vezes ao dia, a mudança do tempo médio do intervalo QTcF, a partir do nível basal, no estado de equilíbrio foi 5 e 8 msec, respectivamente. QTcF de > 500 msec foi observado em 4 pacientes (< 1 % destes pacientes).
Em estudos com voluntários sadios com exposições que foram comparáveis às exposições observadas em pacientes, a mudança no tempo médio do QTcF com placebo a partir do nível basal foi 7 ms (IC ± 4 ms). Nenhum indivíduo teve QTcF > 450 msec. Além disso, nenhuma arritmia clinicamente relevante foi observada durante a condução dos estudos clínicos. Em particular, nenhum episódio de Torsade de pointes (transitórios ou mantidos) foi observado.
O prolongamento clinicamente significativo do intervalo QT pode ocorrer quando Tasigna® é tomado inapropriadamente com alimento e/ou inibidores fortes de CYP3A4 e/ou produtos medicinais com conhecido potencial de prolongar o intervalo QT, portanto, a administração concomitante deve ser evitada.
A presença de hipocalemia e hipomagnesemia pode colocar os pacientes em risco de desenvolver prolongamento do intervalo QT.
Tasigna® deve ser utilizado com cautela em pacientes que têm, ou em risco significativo de desenvolver, o prolongamento do intervalo QTc, tais como:
• síndrome do QT longo;
• doenças cardíacas não controladas ou significativas, incluindo infarto recente do miocárdio, insuficiência cardíaca congestiva, angina instável, ou bradicardia clinicamente significativa.
Este medicamento pode potencializar o prolongamento do intervalo QT, o que aumenta o risco de ataque de arritmias ventriculares graves do tipo “torsades de pointes”, que é potencialmente fatal (morte súbita).
Morte súbita
Nos estudos clínicos, casos incomuns (0,1 a 1%) de morte súbita foram relatados em pacientes com LMC, resistentes ou intolerantes ao imatinibe, em fase crônica ou em fase acelerada recebendo Tasigna® com um histórico médico anterior de doença cardíaca ou fatores de risco cardíaco significativos. Comorbidades em adição à malignidade subjacente também estavam frequentemente presentes, bem como medicações concomitantes. Anomalias de repolarização ventricular podem ter sido fatores de contribuição. Com base na exposição de pós-comercialização em pacientes/ano, a taxa estimada de reportes para relatos espontâneos de morte súbita é de 0,02% por paciente ao ano. Nenhum caso de morte súbita foi reportado no estudo de fase III com LMC Ph+-FC recém-diagnosticado.
Eventos Cardiovasculares
Eventos cardiovasculares foram relatados em um estudo fase III randomizado com nilotinibe em pacientes recémdiagnosticados com LMC, e observados nos relatórios de pós-comercialização. Nos estudos clínicos, com um tempo médio de tratamento de 60,5 meses, eventos cardiovasculares de grau 3/4 incluíram, doença arterial oclusiva periférica (1,4% a 1,1% com 300 mg e 400 mg duas vezes por dia, respectivamente), doença isquêmica cardíaca (2,2% e 6,1%, com 300 mg e 400 mg duas vezes por dia, respectivamente) e eventos isquêmicos cerebrovasculares (1,1% e 2,2% com 300 mg e 400 mg duas vezes por dia, respectivamente). Se ocorrerem sinais ou sintomas agudos de eventos cardiovasculares, aconselhar os pacientes a procurar atendimento médico imediato. O estado cardiovascular dos pacientes deve ser avaliado e os fatores de risco cardiovasculares devem ser monitorados e gerenciados ativamente durante o tratamento com Tasigna® de acordo com as diretrizes padrão.
Retenção de líquidos
Formas graves de retenção de líquidos, tais como derrame pleural, edema pulmonar e derrame pericárdico foram observados pouco frequentemente (0,4 a 1%) em um estudo de Fase III de pacientes recém-diagnosticados com LMC. Eventos similares foram observados em relatórios pós-comercialização. O rápido ganho de peso inesperado deve ser cuidadosamente investigado. Se sinais de retenção grave de líquidos aparecerem durante o tratamento com nilotinibe, a etiologia deve ser avaliada e os doentes tratados em conformidade.
Reativação da hepatite B
A reativação da hepatite B pode ocorrer em pacientes que são portadores crônicos do vírus após receberem um inibidor da tirosina quinase BCR-ABL (ITQ), tal como o nilotinibe. Alguns casos envolvendo medicamentos da classe da ITQ BCR-ABL resultaram em insuficiência hepática aguda ou hepatite fulminante, acarretando em transplante de fígado ou desfecho fatal.
Os pacientes devem ser testados para infecção da hepatite B antes de iniciar o tratamento com Tasigna®. Os pacientes atualmente em uso de Tasigna® devem realizar o teste de valor basal para infecção da hepatite B, a fim de identificar portadores crônicos do vírus. Especialistas em doenças do fígado e no tratamento da hepatite B devem ser consultados antes do tratamento ser iniciado em pacientes com sorologia positiva para hepatite B (incluindo àqueles com doença ativa) e para pacientes cujo teste foi positivo para infecção da hepatite B durante o tratamento. Portadores do vírus da hepatite B que requerem tratamento com Tasigna® devem ser cuidadosamente monitorados para sinais e sintomas de infecção ativa da hepatite B durante o tratamento e por vários meses após a descontinuação do tratamento.
Monitoramento especial de pacientes LMC Ph+-FC que tenham atingido resposta molecular profunda sustentada
Elegibilidade para a Descontinuação do Tratamento
Pacientes elegíveis quanto à expressão de transcrições BCR-ABL típicas, e13a2/b2a2 ou e14a2/b3a2, confirmadas, podem ser considerados para a descontinuação do tratamento. Os pacientes devem apresentar transcrições BCR-ABL típicas para permitir a quantificação dos níveis de BCR-ABL, avaliação da profundidade da reposta molecular, e determinação de perda de remissão molecular após a descontinuação do tratamento com Tasigna®.
Monitoramento de pacientes que tiveram a terapia descontinuada
O monitoramento dos níveis de transcrição BCR-ABL em pacientes elegíveis para a descontinuação de tratamento deve ser realizado com um teste diagnóstico quantitativo validado para medir os níveis de resposta molecular com uma sensibilidade de pelo menos RM 4,5 log (BCR-ABL/ABL ≤ 0,0032% EI). Os níveis de transcrição do gene BCR-ABL devem ser avaliados antes e durante a descontinuação do tratamento.
A perda da resposta molecular maior (RMM = BCR-ABL/ABL ≤ 0,1% EI) ou perda confirmada de RM 4,0 log (duas medidas consecutivas separadas por pelo menos 4 semanas demonstrando perda de RM 4,0 log (BCR-ABL/ABL ≤ 0,01% EI)) acarretará retomada do tratamento dentro de 4 semanas a partir da verificação da ocorrência de perda de remissão. A recaída molecular pode ocorrer durante a fase livre de tratamento, e dados de resultados a longo prazo ainda não estão disponíveis. Portanto, é crucial realizar o monitoramento frequente dos níveis de transcritos BCR ‐ ABL e contagem sanguínea completa com diferencial, para detectar perda de remissão. Para os pacientes que não conseguem atingir a RMM após três meses de reintrodução do tratamento, o teste de mutação no domínio da quinase BCR-ABL deve ser realizado.
Testes laboratoriais e monitoramento
Lipídio sanguíneo
Em estudo de Fase III em pacientes LMC Ph+-FC recém-diagnosticados, 1,1% dos pacientes tratados com 400 mg de nilotinibe duas vezes ao dia apresentaram elevação de colesterol total Grau 3/4; no entanto, não houve elevação de Grau 3/4 no grupo com a dose de 300 mg duas vezes ao dia. Recomenda-se que o perfil lipídico seja avaliado antes de se iniciar o tratamento com Tasigna® e avaliado no 3º e 6º mês após o início da terapia e pelo menos anualmente durante a terapia crônica. Se for necessário o uso de um inibidor da hidroximetilglutaril-CoA (HMG-CoA) redutase (agente redutor de lipídio), consulte a seção “Interações Medicamentosas” antes de iniciar o tratamento, tendo em vista que certos inibidores da HMG-CoA redutase são metabolizados pela via CYP3A4.
Glicose sanguínea
Em estudo de Fase III em pacientes recém-diagnosticados com LMC, 6,9% dos pacientes tratados com 400 mg de nilotinibe duas vezes ao dia apresentaram aumento de glicemia sanguínea grau 3/4; e 7,2% dos pacientes tratados com 300 mg de nilotinibe duas vezes ao dia apresentaram aumento de glicemia grau 3/4. Recomenda-se que o nível de glicose seja avaliado antes do início do tratamento com Tasigna® e monitorado durante o tratamento conforme clinicamente indicado. Se os resultados indicarem necessidade de terapia, o médico deverá seguir tratamento conforme prática clínica.
Interações
A administração de Tasigna® com agentes que são fortes inibidores da CYP3A4 e medicamentos que podem prolongar o intervalo QT tais como antiarrítmicos deve ser evitada. Se o tratamento com qualquer um desses agentes for requerido, recomenda-se, se possível, que a terapia com Tasigna® seja interrompida. Se a interrupção temporária do tratamento com Tasigna® não for possível, indica-se o monitoramento cuidadoso do indivíduo quanto ao prolongamento do intervalo QT.
O uso concomitante de Tasigna® com produtos medicinais que são indutores potentes de CYP3A4 parece reduzir a exposição ao nilotinibe a uma extensão clinicamente relevante. Portanto, em pacientes recebendo Tasigna®, o uso concomitante de agentes terapêuticos alternativos com menor potencial para indução de CYP3A4 deve ser selecionado.
Interação com alimentos
A biodisponibilidade do nilotinibe é aumentada pelos alimentos. Tasigna® não deve ser tomado junto com alimentos e deve ser ingerido duas horas após a refeição. Nenhum alimento deve ser consumido por pelo menos uma hora após a ingestão da dose.
Suco de grapefruit e outros alimentos que sejam conhecidos por inibir a CYP3A4 devem ser evitados em qualquer momento.
Insuficiência hepática
A insuficiência hepática tem um efeito modesto na farmacocinética de nilotinibe. A administração de uma dose única de nilotinibe resultou em aumentos na AUC de 35%, 35% e 19% em pacientes com insuficiência hepática leve, moderada e grave respectivamente, comparado ao grupo controle de indivíduos com função hepática normal. A Cmáx prevista no estado de equilíbrio de nilotinibe mostrou aumentos de 29%, 18% e 22%, respectivamente. Os estudos clínicos excluíram pacientes com alanina aminotransferase (ALT) e/ou aspartato aminotransferase (AST) > 2,5 (ou > 5, se relacionada à doença) vezes o limite superior da normalidade e/ou bilirrubina total > 1,5 vezes o limite superior da normalidade. O metabolismo do nilotinibe é principalmente hepático. Recomenda-se cautela em pacientes com insuficiência hepática.
Este medicamento pode causar hepatotoxicidade. Por isso, requer uso cuidadoso, sob vigilância médica estrita e acompanhado por controles periódicos da função hepática mensalmente ou conforme clinicamente indicado.
Lipase sérica
Foi observada elevação na lipase sérica. Recomenda-se cautela em pacientes com história prévia de pancreatite. Caso as elevações de lipase sejam acompanhadas por sintomas abdominais, a dose deve ser interrompida e um diagnóstico apropriado deve ser considerado para excluir a pancreatite.
Gastrectomia total
A biodisponibilidade de nilotinibe pode ser reduzida em pacientes com gastrectomia total. Deve-se considerar acompanhamento mais frequente destes pacientes.
Síndrome de lise tumoral (SLT)
Casos de SLT foram relatados em pacientes tratados com Tasigna®.
Lactose
Como as cápsulas contêm lactose, Tasigna® não é recomendado para pacientes com problemas hereditários raros de intolerância à galactose, deficiência grave de lactase, deficiência ou má absorção de glucose-galactose.
Este medicamento não deve ser usado por pessoas com síndrome de má-absorção de glicose-galactose.
Gravidez, lactação, mulheres e homens com potencial reprodutivo
Gravidez
Tasigna® pode causar dano fetal quando administrado a uma mulher grávida. Não há dados adequados sobre o uso de Tasigna® em mulheres grávidas. Estudos reprodutivos em ratos e coelhos demonstraram que o nilotinibe induziu toxicidade embrionária e/ou toxicidade do feto (após a exposição pré-natal ao nilotinibe) em exposições iguais às obtidas em seres humanos na dose máxima recomendada humana de 400mg duas vezes ao dia. Tasigna® não deve ser utilizado durante a gravidez, a menos que seja necessário. Se for utilizado durante a gravidez ou a paciente ficar grávida durante o tratamento com Tasigna®, a paciente deve ser informada sobre os riscos potenciais para o feto.
Existe uma quantidade limitada de dados sobre gravidezes em pacientes durante a tentativa de remissão livre de tratamento (TFR). Se a gravidez ocorrer durante a fase de TFR, a paciente deve ser informada sobre a necessidade potencial de reiniciar o tratamento com Tasigna® durante a gravidez.
Tasigna® enquadra-se na categoria D de risco na gravidez.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. Informe imediatamente seu médico em caso de suspeita de gravidez.
Dados em animais
O nilotinibe não induziu teratogenicidade, mas mostrou toxicidade embrionária e feto em doses que também apresentaram toxidade materna. O aumento do aborto pós-implantação foi observado tanto no estudo de fertilidade quanto no tratamento de machos e fêmeas e no estudo de toxicidade embrionária com o tratamento de fêmeas. A letalidade de embriões e os efeitos do feto (principalmente diminuição do peso fetal, variações viscerais e esqueléticas) em ratos e aumento da reabsorção de fetos e variações esqueléticas em coelhos estavam presentes nos estudos de toxicidade embrionária. A exposição ao nilotinibe em fêmeas com níveis de efeitos adversos não observados (NEANO) foi geralmente menor ou igual à dos humanos a 800 mg/dia. Em um estudo pré e pós-natal, a administração oral de nilotinibe a ratos fêmeas a partir do dia 6 da gestação até o dia 21 ou 22 pós-parto resultou em efeitos maternos (redução do consumo de alimentos e menores ganhos de peso corporal) e maior período de gestação a 60mg/kg. A dose materna de 60mg/kg foi associada a diminuição do peso corporal do filhote e mudanças em alguns parâmetros de desenvolvimento físico (média de dias para abertura do pavilhão auricular, erupção dentária e abertura do olho foi prematura). O NEANO em animais maternos e descendentes foi uma dose materna de 20mg/kg.
Lactação
Não é conhecido se o nilotinibe é excretado no leite humano. Estudos em animais demonstram que o nilotinibe é excretado no leite. Lactentes não devem amamentar enquanto estiverem tomando Tasigna® e por 2 semanas após a última dose, pois um risco para o recém-nascido não pode ser excluído.
Uso contraindicado no aleitamento ou na doação de leite humano.
Contracepção
Mulheres com o potencial reprodutivo devem ser aconselhadas a usar um método de contracepção eficaz (métodos que resultam em taxas de gravidez inferiores a 1%) ao receber Tasigna® e por até 2 semanas após o término do tratamento com Tasigna®.
Infertilidade
O efeito do nilotinibe sobre a fertilidade masculina e feminina não é conhecido. De acordo com estudos realizados em animais, nenhum efeito foi observado na contagem/motilidade dos espermatozoides e na fertilidade em ratos machos e fêmeas até a dose mais alta testada de aproximadamente 5 vezes maior do que a dosagem recomendada para humanos.
Efeitos na habilidade de dirigir e utilizar máquinas
Nenhum estudo sobre os efeitos do nilotinibe na habilidade de dirigir e operar máquinas foi realizado. Pacientes que sentirem tontura, distúrbio visual ou outras reações adversas com um impacto potencial na habilidade de dirigir ou de operar máquinas com segurança, não devem realizar estas atividades enquanto persistam as reações adversas.
Atenção: Contém os corantes dióxido de titânio, óxido de ferro amarelo e óxido de ferro vermelho (Tasigna® 150 mg) que podem, eventualmente, causar reações alérgicas.
Interações medicamentosas
O nilotinibe é metabolizado principalmente pelo fígado sendo a CYP3A4 a principal contribuinte para o metabolismo oxidativo. Nilotinibe é também é um substrato para a bomba de efluxo multidrogas da glicoproteína-P (P-gp). Portanto, a absorção e a subsequente eliminação do nilotinibe absorvido via sistêmica podem ser influenciadas por medicamentos que afetam a CYP3A4 e/ou a P-gp.
Medicamentos que podem aumentar as concentrações plasmáticas de nilotinibe
Em um estudo de Fase I de nilotinibe administrado em combinação com imatinibe (um substrato e moderador de Pgp e CYP3A4), ambos os medicamentos tiveram um efeito inibitório leve na CYP3A4 e/ou Pgp. Quando os dois medicamentos foram administrados concomitantemente, a AUC de imatinibe foi aumentada em 18% a 39%, e a AUC de nilotinibe foi aumentada em 18% a 40%.
A biodisponibilidade do nilotinibe em indivíduos sadios foi aumentada em 3 vezes quando coadministrado com um forte inibidor da CYP3A4, o cetoconazol. Portanto, o tratamento concomitante com fortes inibidores da CYP3A4 deve ser evitado (incluindo, mas não limitado a cetoconazol, itraconazol, voriconazol, ritonavir, claritromicina e telitromicina). Medicações concomitantes alternativas, com nenhuma ou com uma mínima inibição da CYP3A4, devem ser consideradas.
Medicamentos que podem diminuir as concentrações plasmáticas de nilotinibe
Em voluntários sadios recebendo um indutor de CYP3A4, a rifampicina, na dose de 600 mg por dia por 12 dias, a exposição sistêmica (AUC) ao nilotinibe foi diminuída aproximadamente em 80%.
Indutores de atividade da CYP3A4 podem aumentar o metabolismo do nilotinibe e, com isso, diminuir as concentrações plasmáticas de nilotinibe. A administração concomitante de medicamentos que induzem a CYP3A4 (por ex.: fenitoína, rifampicina, carbamazepina, fenobarbital, e erva de São João) pode reduzir a exposição ao nilotinibe. Em pacientes para os quais indutores da CYP3A4 são indicados, agentes alternativos com um menor potencial de indução da enzima devem ser considerados.
O nilotinibe tem uma solubilidade pH dependente, com solubilidade mais baixa em pH mais alto. Em indivíduos sadios recebendo 40 mg de esomeprazol uma vez ao dia por 5 dias, o pH gástrico foi aumentado notavelmente, mas a absorção de nilotinibe foi diminuída modestamente (diminuição de 27% na Cmáx e 34% na AUC0-∞). O nilotinibe tem uma solubilidade dependente do pH, apresentando uma diminuição da solubilidade em pH mais elevado. Medicamentos, tais como inibidores da bomba de prótons que inibem a secreção de ácido gástrico para elevar o pH gástrico, podem diminuir a solubilidade do nilotinibe e reduzir sua biodisponibilidade. Em indivíduos saudáveis, a coadministração de uma dose única de 400 mg de Tasigna® com doses múltiplas de esomeprazol (um inibidor da bomba de prótons) a 40 mg por dia diminuiu a AUC do nilotinibe em 34%. O aumento da dose de Tasigna® quando coadministrado com esses medicamentos provavelmente não compensa pela perda de exposição. Como os inibidores da bomba de prótons afetam o pH do trato gastrointestinal superior por um período prolongado, a separação de doses pode não eliminar a interação.
O uso concomitante de inibidores da bomba de prótons com Tasigna® não é recomendado.
Em estudo realizado com indivíduos sadios, não foi observada mudança significativa na farmacocinética de nilotinibe após administração de dose única de 400 mg de Tasigna® administrada 10 horas após e 2 horas antes de famotidina.
Portanto, quando é necessária a administração concomitante de um bloqueador H2, este deve ser administrado aproximadamente 10 horas antes e cerca de 2 horas após a dose de Tasigna®. No mesmo estudo mencionado acima, a administração de antiácidos (hidróxido de alumínio/hidróxido de magnésio/simeticona) 2 horas antes ou após uma dose única de 400 mg de Tasigna® não alterou a farmacocinética de nilotinibe. Portanto, se necessário, um antiácido pode ser administrado cerca de 2 horas antes ou 2 horas após a dose de Tasigna®.
Medicamentos cuja concentração sistêmica pode ser alterada pelo nilotinibe O nilotinibe é identificado como um inibidor competitivo da CYP3A4, CYP2C8, CYP2C9, CYP2D6 e da UGT1A1, com o valor de Ki sendo mais baixo para CYP2C9 (Ki = 0,13 microM). Os estudos de indução de enzimas indicam que o nilotinibe pode ser considerado um indutor in vitro das atividades de CYP2B6, CYP2C8 e CYP2C9.
Em pacientes com LMC, a administração de 400 mg de nilotinibe duas vezes por dia durante 12 dias aumentou a exposição sistêmica do midazolam (um substrato da CYP3A4) administrado por via oral em 2,6 vezes. O nilotinibe é um inibidor moderado da CYP3A4. Como resultado, a exposição sistêmica de outros medicamentos metabolizados principalmente pela CYP3A4 (por exemplo, certos inibidores da HMG-CoA redutase) pode ser aumentada quando coadministrados com nilotinibe. O monitoramento e o ajuste de dose adequados podem ser necessários para as drogas que são substratos da CYP3A4 e que possuem um índice terapêutico estreito (incluindo, mas não se limitando a alfentanil, ciclosporina, di-hidroergotamina, ergotamina, fentanil, sirolimo e tacrolimo), quando coadministradas com nilotinibe.
Em indivíduos sadios, nas concentrações clinicamente relevantes de nilotinibe, a farmacocinética ou farmacodinâmica da varfarina, um substrato sensível da CYP2C9, não foi alterada. Tasigna® pode ser usado concomitantemente com varfarina sem aumentar o efeito anticoagulante.
Medicamentos antiarrítmicos e outras drogas que podem prolongar o intervalo QT
O uso concomitante de medicamentos antiarrítmicos (incluindo, mas não limitado à amiodarona, disopiramida, procainamida, quinidina e sotalol) e outras drogas que podem prolongar o intervalo QT (incluindo, mas não limitado à cloroquina, halofantrina, claritromicina, haloperidol, metadona, moxifloxacino, bepridil e pimozida) deve ser evitado.
Interações com alimentos
A absorção e a biodisponibilidade de nilotinibe são aumentadas quando administrado com alimentos, resultando em concentração sérica mais alta. Suco de grapefruit e outros alimentos conhecidos por inibir a CYP3A4 devem ser evitados a qualquer momento.
Estabilidade/ Conservação
Este medicamento deve ser armazenado em temperatura ambiente (entre 15 e 30 °C), e depois de aberto mantido na embalagem original.
Reações adversas
Resumo do perfil de segurança
O perfil de segurança do nilotinibe, descrito abaixo, é baseado em dados de pacientes recém-diagnosticados com LMC Ph+-FC em um estudo de fase III, randomizado, aberto, controlado com comparador ativo e um estudo com pacientes com LMC Ph+-FC e LMC-FA resistentes ou intolerantes, que serviu de base para as indicações listadas. Informações de segurança também são fornecidas a partir de dois estudos de descontinuação do tratamento com Tasigna® e de um estudo fase III em pacientes com LMC Ph+ em fase crônica com respota sub-ótima a imatinibe.
Em pacientes com LMC Ph+-FC recém-diagnosticada
Os dados reportados abaixo refletem a exposição a Tasigna em um estudo randomizado de fase III em pacientes com LMC Ph+ em fase crônica recém-diagnosticada tratados na dose recomendada de 300 mg duas vezes ao dia (n = 279). O tempo médio em tratamento foi de 60,5 meses (variando de 0,1 a 70,8 meses).
As reações adversas não hematológicas relacionadas ao medicamento relatadas como muito comuns (≥ 10%) foram rash (erupção cutânea), prurido, cefaleia, náusea, fadiga, alopecia, mialgia e dor abdominal superior. A maioria destas reações adversas foi de gravidade leve a moderada (Graus 1 ou 2). Constipação, diarreia, pele seca, cãimbras musculares, artralgia, dor abdominal, edema periférico, vômito e astenia foram menos comumente observados (< 10% e ≥ 5%) e foram de gravidade leve a moderada, clinicamente manejáveis e geralmente não necessitaram de redução de dose. Derrames pleurais e pericárdicos, independentemente da causalidade, ocorreram em 2% e < 1% dos pacientes, respectivamente, recebendo Tasigna 300 mg duas vezes ao dia. Hemorragia gastrintestinal, independente da causalidade, foi relatada em 3% desses pacientes.
A alteração no tempo médio do intervalo QTcF em relação ao valor basal no estado de equilíbrio na dose recomendada de 300 mg de nilotinibe duas vezes ao dia foi 6 ms. No grupo de nilotinibe 400 mg duas vezes ao dia e no grupo de imatinibe 400 mg uma vez ao dia, a alteração média em relação ao valor basal do tempo médio do intervalo QTcF no estado de equilíbrio foi de 6 ms e 3 ms, respectivamente. Nenhum paciente apresentou QTcF absoluto > 500 msec em quaisquer dos grupos de tratamento com Tasigna® e nenhum evento de Torsade de Pointes foi observado. O aumento QTcF a partir do nível basal que excedeu 60 ms foi observado em 5 pacientes dos grupos tratados com Tasigna® (um no grupo de tratamento de 300 mg duas vezes ao dia e quatros no grupo de tratamento com 400 mg duas vezes ao dia).
Após a análise de 60 meses, não houve pacientes em quaisquer grupos de tratamento com fração de ejeção do ventrículo esquerdo (FEVE) < 45% durante o tratamento. Também não houve pacientes com redução de 15% ou mais na FEVE a partir do nível basal.
Nenhuma morte súbita foi relatada em qualquer grupo de tratamento.
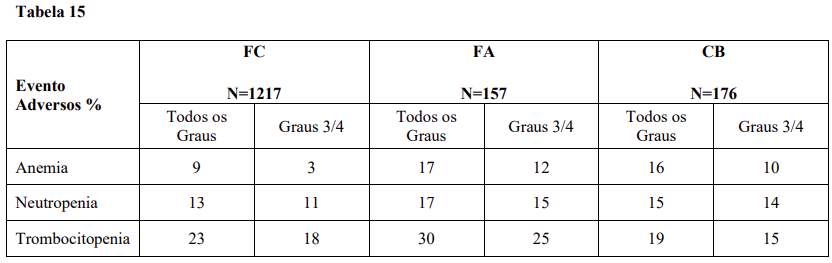
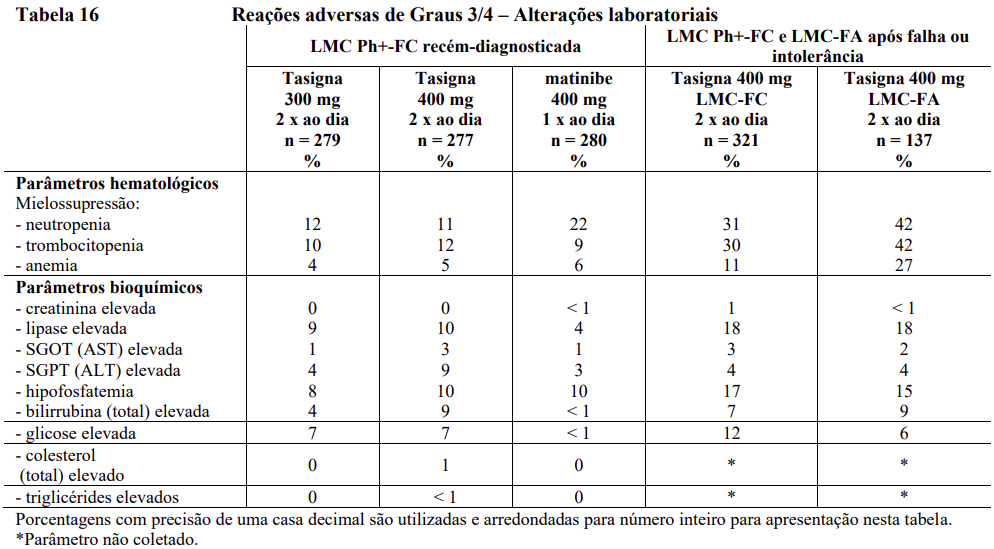
No grupo do nilotinibe de 300 mg duas vezes ao dia, reações adversas hematológicas incluem mielossupressão: trombocitopenia (18%), neutropenia (15%), e anemia (8%). Reações adversas bioquímicas incluem aumento da alanina aminotransferase (24%), hiperbilirrubinemia (16%), aumento da aspartato aminotransferase (12%), aumento de lipase (11%), aumento de bilirrubina sanguínea (10%), hiperglicemia (4%), hipercolesterolemia (3%) e hipertrigliceridemia (< 1%). Vide Tabela 15 para anormalidades laboratoriais de Graus 3 e 4.
A descontinuação devido a reações adversas relacionadas ao medicamento foi observada em 10% dos pacientes.
Em pacientes com LMC Ph+-FC e LMC-FA após falha ou intolerância
Os dados descritos abaixo refletem a exposição ao Tasigna® em 458 pacientes com LMC Ph+-FC (n=321) e LMC- Ph+ FA (n=137) após falha ou intolerância a pelo menos uma terapia prévia, incluindo imatinibe em um estudo multicêntrico aberto tratado com a dose recomendada de 400 mg duas vezes ao dia. As reações adversas não hematológicas relacionadas ao medicamento relatadas como muito comuns (≥ 10% nas populações de pacientes com LMC-FC e LMC-FA combinada) foram: rash (erupção cutânea), prurido, náusea, fadiga, cefaleia, constipação, diarreia, vomito e mialgia. A maioria destas reações adversas foi de gravidade leve a moderada. Alopecia, espasmos musculares, diminuição de apetite, artralgia, dor óssea, dor abdominal, edema periférico e astenia foram menos frequentemente observados (< 10% e ≥ 5%) e tiveram gravidade leve a moderada (Grau 1 ou 2).
Derrames pleurais e pericárdicos, assim como, complicações devido à retenção de líquidos ocorreram em < 1% dos pacientes que receberam Tasigna®. Insuficiência cardíaca foi observada em < 1% dos pacientes. Hemorragias gastrointestinais e de sistema nervoso central (SNC) foram relatadas em 1% e < 1% dos pacientes, respectivamente. QTcF excedendo 500 ms foi observado nesse estudo em 4 pacientes (< 1%). Nenhum episódio de Torsade de Pointes (transitório ou mantido) foi observado.
Reações adversas hematológicas incluem mielossupressão: trombocitopenia (31%), neutropenia (17%) e anemia (14%). Vide Tabela 15 – Reações adversas de Graus 3/4 – Alterações laboratoriais.
A descontinuação devido a reações adversas relacionadas ao medicamento foi observada em 16% dos pacientes em FC e 10% dos pacientes em FA.
Em pacientes com LMC Ph+-FC que não tenham atingido resposta molecular maior ou igual a uma redução 4.5 log em tratamento com imatinibe
Os dados relatados abaixo foram obtidos em um estudo fase III, randomizado, aberto, no qual pacientes adultos, tanto homens quanto mulheres, diagnosticados com LMC Ph+-FC e após 2 anos de terapia com imatinibe foram expostos a Tasigna 400 mg uma vez ao dia versus imatinibe 400 mg ou 600 mg uma vez ao dia por 48 meses. Os pacientes randomizados no braço de imatinibe receberam a mesma dose de imatinibe como antes da randomização. A duração média de exposição foi 47,2 meses no braço de Tasigna® e 37,0 meses e 26,7 meses nas coortes com doses de 400 mg e 600 mg no braço de imatinibe, respectivamente.
As reações adversas relacionadas ao medicamento relatadas por, pelo menos, 20% dos pacientes no grupo de Tasigna® e mais frequentemente comparada ao grupo de imatinibe foram cefaleia, rash (erupção cutânea) e prurido. Uma maior proporção de pacientes no grupo de Tasigna® relatou EAs levando à descontinuação e EAs exigindo ajuste de dose/ interrupção comparada àqueles no grupo de imatinibe. Aumento na bilirrubina e transaminases foi comumente relatado após tratamento com Tasigna®.
Até a data de corte de 48 meses, três mortes durante o tratamento foram relatadas (duas no braço de Tasigna® e uma no braço de imatinibe). Três pacientes morreram mais de 28 dias após descontinuação do medicamento em estudo (um no braço de Tasigna® e dois no braço de imatinibe).
Intervalos QTc > 450 ms foram observados em 4 pacientes recebendo terapia com Tasigna® no oitavo dia. Nenhum paciente apresentou intervalo QTc > 480 ms. Aumento no intervalo QTc > 30 ms em relação ao valor basal foi reportado para 8 pacientes (7,9%). Nenhum paciente vivenciou prolongamento do intervalo QTc > 60 ms no grupo de Tasigna®.
Reações adversas mais frequentemente relatadas
As reações adversas não hematológicas (excluindo anormalidades laboratoriais), que foram relatadas em pelo menos 5% dos pacientes, em qualquer um dos estudos clínicos com Tasigna®, que servem como base para as indicações listadas, estão apresentadas na Tabela 13. As reações adversas estão classificadas por ordem de frequência, a mais frequente primeiro. Dentro de cada grupo de frequência, as reações adversas são apresentadas em ordem decrescente de gravidade. Adicionalmente, a categoria de frequência correspondente para cada reação adversa é baseada na seguinte convenção: muito comum (≥ 1/10) ou comum (≥ 1/100 a < 1/10). A frequência é baseada num valor mais alto para qualquer grupo de Tasigna® nos dois estudos, utilizando uma precisão decimal para porcentagens.
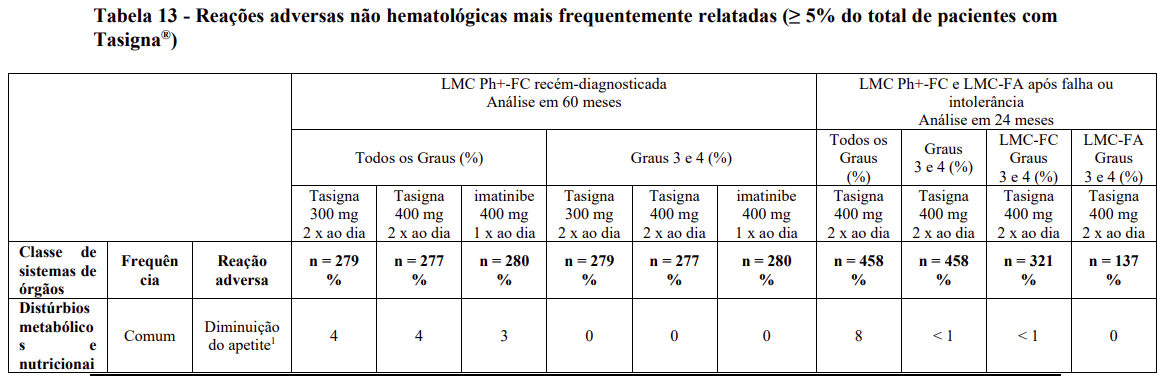
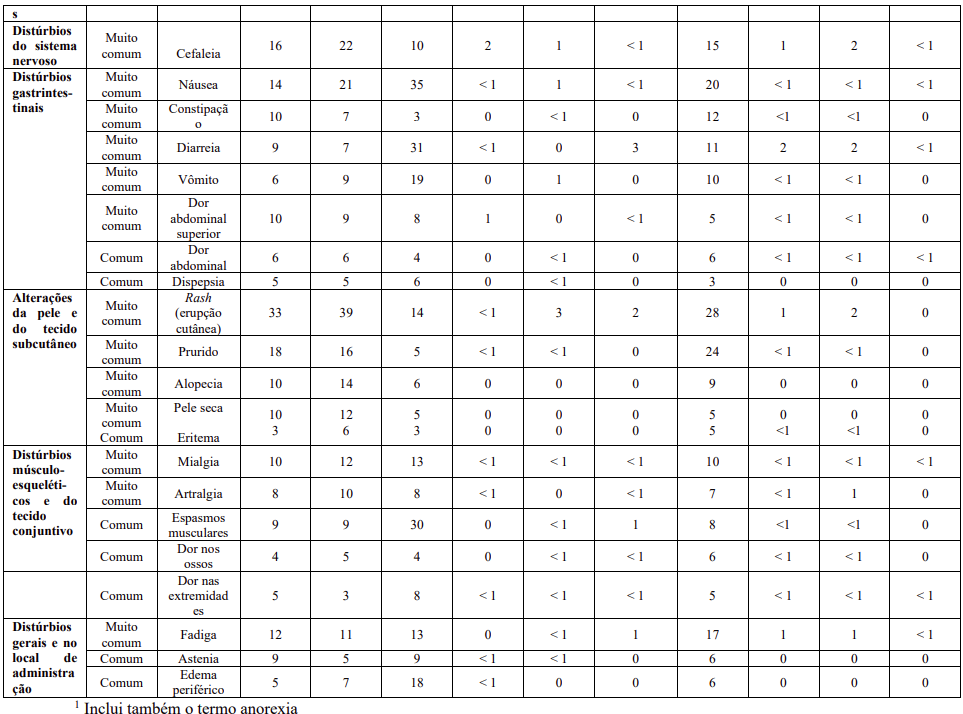
As porcentagens são arredondadas para um número inteiro para apresentação nesta tabela. No entanto, as porcentagens com uma casa decimal são usadas para identificar os termos com frequência de pelo menos 5% e para classificar os termos de acordo com as categorias de frequência.
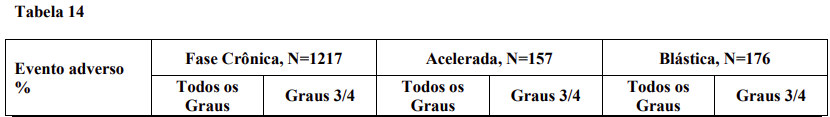
Adicionalmente, o estudo 2109, um estudo fase 3, aberto, multicêntrico, internacional, cujo objetivo primário foi o de confirmar o perfil de segurança do nilotinibe numa população maior de pacientes está relatado abaixo. O estudo 2109 envolveu pacientes adultos com LMC em fase crônica, fase acelerada e crise blástica, resistentes ou intolerantes ao imatinibe.
As medianas de duração de exposição ao nilotinibe foram de 184 dias para fase crônica, 17 dias para fase acelerada e 62 dias para pacientes em crise blástica. A dose mediana recebida foi de 792 mg, 787 mg e 783 mg diários respectivamente. A redução de dose do nilotinibe devido a eventos adversos ocorreu em 20% dos pacientes em fase crônica e em 22% dos pacientes em fase acelerada e em crise blástica. Entretanto, a descontinuação do tratamento devido a eventos adversos foi observada em apenas 12% dos pacientes em fase crônica e em 13% e 11% dos pacientes em fase acelerada e crise blástica, respectivamente.
A tabela 14 descreve os eventos adversos não hematológicos mais frequentemente observados neste estudo.
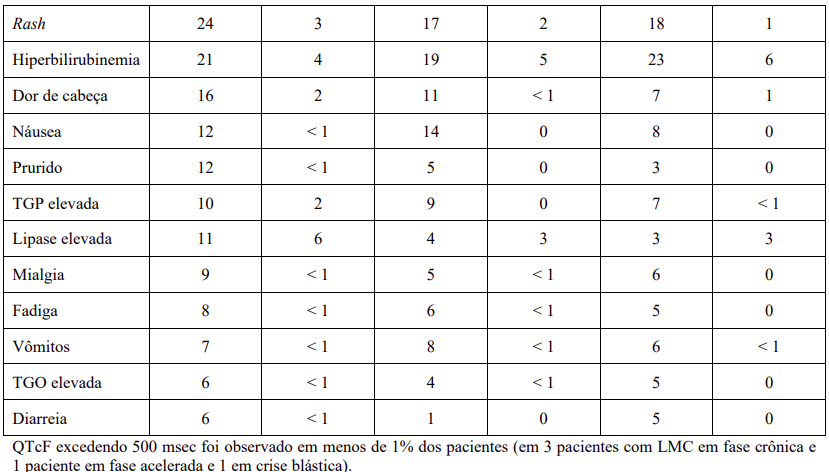
A tabela 15 sumariza os eventos adversos hematológicos observados.
Dados adicionais dos estudos clínicos
As seguintes reações adversas foram relatadas em pacientes dos estudos clínicos com Tasigna®, que servem como base para as indicações listadas, na dose recomendada com uma frequência menor que 5% (comuns ≥ 1/100 a < 1/10; incomuns ≥ 1/1000 a < 1/100; eventos únicos são capturados como de frequência desconhecida). Para anormalidades laboratoriais, eventos muitos comuns (> 1/10) não incluídos na Tabela 13 também são relatados. As reações adversas foram incluídas com base na relevância clínica e classificadas em ordem decrescente de gravidade dentro de cada categoria, obtidas a partir de dois estudos clínicos: 1-) análise de 60 meses para Ph+ LMC-FC recém diagnosticada; 2-) análise de 24 meses para LMC Ph+-FC e LMC-FA resistente ou intolerante.

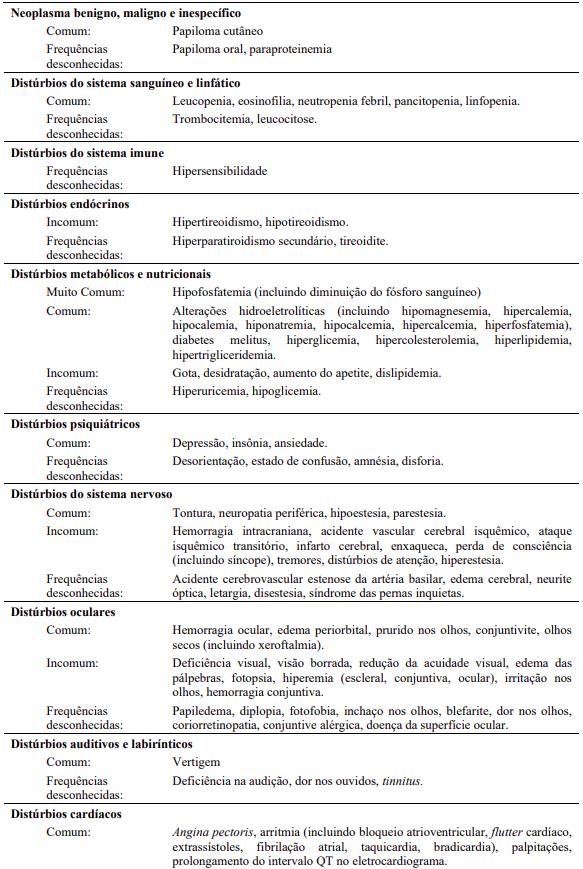
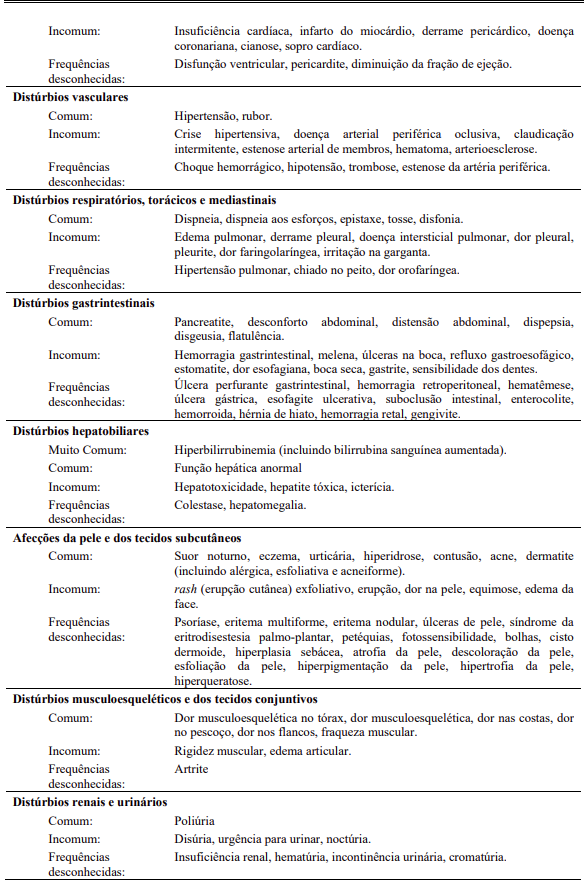
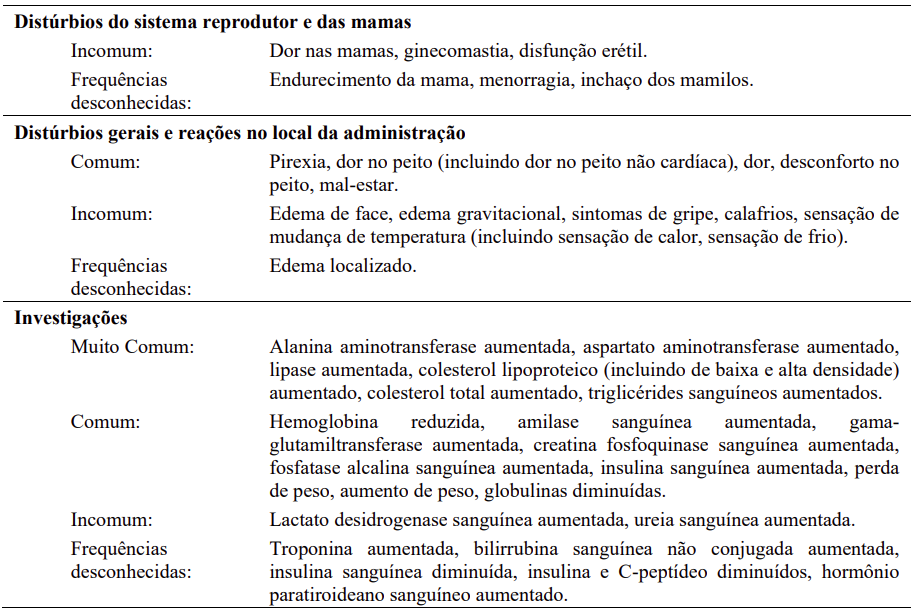
Anormalidades laboratoriais
Alterações clinicamente relevantes ou graves nos valores laboratoriais hematológicos ou bioquímicos de rotina estão apresentadas na Tabela 16.
Descontinuação do tratamento em pacientes com LMC Ph+-FC que tenham atingido resposta molecular profunda sustentada
Após a descontinuação do tratamento com Tasigna® no âmbito de tentativa de remissão livre de tratamento (TFR), sintomas musculoesqueléticos (por exemplo, mialgia, dores nas extremidades, artralgia, dor óssea, dor na coluna ou dor musculoesquelética), foram relatados com mais frequência do que antes da descontinuação do tratamento no primeiro ano, conforme observado na Tabela 17. A taxa de novos sintomas osteomusculares geralmente diminuiu no segundo ano após a descontinuação do tratamento.
Em um estudo clínico de fase II com pacientes com LMC Ph+-FC recém-diagnosticada (n=190), os sintomas musculoesqueléticos foram relatados com mais frequência do que antes da descontinuação do tratamento no primeiro ano, enquanto a taxa de novos eventos geralmente diminuiu no segundo ano após a descontinuação do tratamento e continua a diminuir ao longo dos anos até o terceiro ano após a descontinuação (16,0% no ano antes da descontinuação e 40,4%, 9,6%, 4,3% do primeiro ao terceiro ano após a descontinuação do nilotinibe).
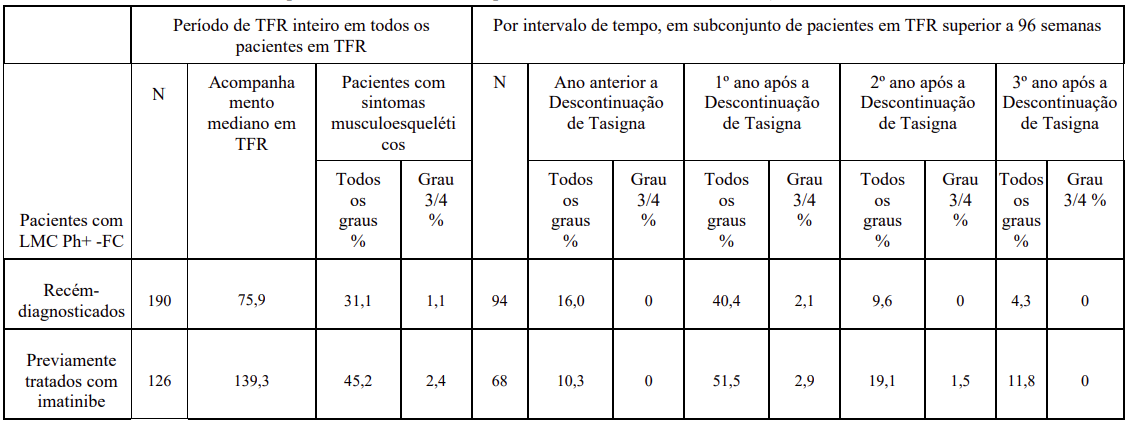
Em um estudo clínico de fase II com pacientes com LMC Ph+-FC em tratamento com Tasigna® e previamente tratados com imatinibe (n=126), sintomas musculoesqueléticos dentro de um ano da descontinuação foram reportados em 42,1% versus 14,3% dos anos anteriores, durante o tratamento com Tasigna®.
Tabela 17 Sintomas musculoesqueléticos que ocorrem com a descontinuação do tratamento no contexto de remissão livre de tratamento (TFR)
Reações adversas de casos de relatos espontâneos e da literatura (frequência desconhecida)
As seguintes reações adversas foram derivadas de experiência pós-comercialização com Tasigna® via relatos de casos espontâneos, casos da literatura, programas de acesso expandido, e outros estudos clínicos além dos estudos globais de registro. Como estas reações foram relatadas voluntariamente a partir de uma população de tamanho incerto, não é sempre possível ter uma estimativa confiável de sua frequência ou estabelecer uma relação causal a exposição ao nilotinibe. Frequência desconhecida: síndrome da lise tumoral, paralisia facial.
Contraindicações
Tasigna® é contraindicado para pacientes com hipersensibilidade conhecida ao nilotinibe ou a qualquer excipiente do produto.
Fonte:
Tasigna®. [Bula]. São Paulo: Novartis Biociências S.A. Disponível em: https://consultas.anvisa.gov.br/#/bulario/q/?numeroRegistro=100681060: 15/08/2025