Nome Comercial/ Apresentação
Tysabri (natalizumabe) 20 mg/mL / Frasco-ampola
Classe Terapêutica
Anticorpo monoclonal
Indicação
TYSABRI™ (natalizumabe) é indicado como terapia única no tratamento da Esclerose Múltipla recorrente-remitente, para prevenir e retardar a progressão da incapacidade nos seguintes grupos de pacientes:
• Pacientes que não responderam a um ciclo completo e adequado com outros medicamentos. Os pacientes devem ter apresentado pelo menos 1 recidiva no ano anterior durante o tratamento e ter pelo menos 9 lesões T2 hiperintensas na Ressonância Magnética Nuclear (RMN) craniana ou pelo menos 1 lesão realçada por gadolínio. ou
• Pacientes com Esclerose Múltipla recorrente-remitente grave em rápida evolução, definida por 2 ou mais recidivas incapacitantes no espaço de um ano e com 1 ou mais lesões realçadas por gadolínio em uma imagem do cérebro obtida por Ressonância Magnética Nuclear (RMN) ou um aumento significativo das lesões em T2 comparativamente com uma RMN anterior recente.
Dose
O tratamento deve ser iniciado e continuamente supervisionado por um médico especialista com experiência em diagnóstico e tratamento de doenças neurológicas, em centros com fácil acesso a imagens de Ressonância Magnética Nuclear (RMN).
Após 2 anos de tratamento, os pacientes deverão ser informados novamente sobre os riscos, especialmente em relação ao aumento do risco de desenvolvimento de Leucoencefalopatia Multifocal Progressiva (LMP), e devem ser instruídos, junto com seus cuidadores, sobre os sinais e sintomas da LMP.
Os pacientes podem passar diretamente do tratamento com betainterferonas ou acetato de glatirâmer para o natalizumabe desde que não existam anomalias importantes relacionadas com o tratamento anterior como, por exemplo, neutropenia. Se existirem sinais de anomalias relacionadas com o tratamento anterior, estes devem voltar ao normal antes de se dar início ao tratamento.
É possível que alguns pacientes tenham sido expostos a medicamentos imunossupressores como, por exemplo, mitoxantrona, ciclofosfamida e azatioprina. Estes fármacos têm o efeito potencial de provocar imunossupressão prolongada, mesmo depois de ter sido suspenso o tratamento. Por este motivo, antes de iniciar o tratamento com natalizumabe, o médico deve confirmar se estes pacientes não se encontram imunocomprometidos.
Ajuste de dose
Insuficiência renal: não há ajustes de dosagem previstos na bula do fabricante.
Insuficiência hepática: não há ajustes de dosagem previstos na bula do fabricante.
Preparo
Natalizumabe apresenta-se sob a forma de solução concentrada que deve ser diluída em solução de cloreto de sódio a 9 mg/mL (0,9%) antes da administração por infusão intravenosa. Após a diluição, a infusão deve ser administrada por aproximadamente 1 hora.
Vias de administração
INTRAVENOSA
Administração
TYSABRI™ (natalizumabe) não deve ser administrado como injeção em bolus.
Instruções de uso:
1. Antes da diluição e administração, inspecione o frasco-ampola quanto a presença de partículas. Se forem observadas partículas e/ou se o líquido no frasco-ampola não estiver incolor, de transparente a levemente opalescente, o medicamento não deverá ser utilizado.
2. Utilize técnica asséptica ao preparar a solução para infusão intravenosa (IV). Retire a tampa flip-off do frasco-ampola. Introduza a agulha da seringa no frasco-ampola no centro da rolha de borracha e retire os 15 mL de solução concentrada.
3. Adicione os 15 mL de solução concentrada a 100 mL de solução de cloreto de sódio a 9 mg/ml (0,9%). Inverta cuidadosamente a solução de modo a misturar completamente. Não agitar.
4. Natalizumabe não deve ser misturado com outros medicamentos ou diluentes.
5. Inspecione visualmente o produto diluído para verificar se apresenta partículas ou descoloração antes da administração. Não utilize se observar descoloração ou partículas estranhas.
6. O produto diluído deve ser utilizado o mais rapidamente possível e sempre nas 8 horas seguintes à diluição. Se o produto diluído for conservado a uma temperatura entre 2˚C e 8˚C (não congelar), deixe a solução atingir a temperatura ambiente antes da infusão.
7. A solução diluída deve ser administrada por infusão intravenosa ao longo de 1 hora a uma velocidade de aproximadamente 2 mL/minuto.
8. Após a conclusão da infusão, lave a linha intravenosa com uma solução de cloreto de sódio a 9 mg/ml (0,9%).
9. Cada frasco-ampola destina-se apenas a uma única utilização.
10. Os produtos não utilizados ou os resíduos da infusão devem ser eliminados adequadamente.
Cuidados específicos e monitoramento
Alguns pacientes apresentaram reações de hipersensibilidade. Por isso, recomenda-se que o paciente fique em observação durante a infusão e durante a hora seguinte ao procedimento.
Todos os recursos para o tratamento de hipersensibilidade devem estar disponíveis no local da infusão, bem como o acesso a imagens de Ressonância Magnética Nuclear.
Após as primeiras 12 doses intravenosas, os pacientes devem continuar a ser observados durante a infusão e, após a infusão, devem ser observados de acordo com o julgamento clínico.
Interações medicamentosas
O uso em combinação com betainterferonas e acetato de glatirâmer está contraindicado.
Imunização
Em um estudo randomizado e aberto com 60 pacientes portadores de EM recorrente-remitente, não houve diferença significativa na resposta imune humoral a uma reexposição ao antígeno (toxoide tetânico) e somente uma resposta imune humoral um pouco mais lenta e reduzida a um neoantígeno (hemocianina ou KLH – keyhole limpet haemocyanin) foi observada em pacientes que foram tratados com natalizumabe por 6 meses comparado com o grupo controle não tratado. Vacinas vivas não foram estudadas.
Estabilidade/ Conservação
Deve ser conservado sob refrigeração (2ºC a 8ºC). Não congelar. Mantenha o frasco-ampola dentro da embalagem para protegê-lo da luz
Reações adversas
Uma vez que estudos clínicos são conduzidos em diversas situações, as taxas de reações adversas observadas nos estudos de um medicamento não podem ser diretamente comparadas às taxas de estudos clínicos de outro medicamento e podem não corresponder às taxas observadas na prática clínica.
As reações adversas mais comuns (incidência ≥ 10%) foram dor de cabeça e fadiga em ambos os estudos conduzidos com população de pacientes portadores de EM e de pacientes não portadores de EM. Outras reações adversas comuns (incidência ≥ 10%) na população de pacientes com EM foram artralgia, infecção do trato urinário, infecção do trato respiratório inferior, gastroenterite, vaginite, depressão, dor nas extremidades, desconforto abdominal, diarreia não especificada e erupções cutâneas. Outras reações adversas comuns (incidência ≥ 10%) em pacientes não portadores de EM foram infecções do trato respiratório superior e náusea.
Em estudos controlados por placebo realizados em 1.617 pacientes com EM tratados com natalizumabe por no máximo 2 anos (placebo: 1.135), ocorreram reações adversas que levaram à interrupção do tratamento em 5,8% dos pacientes tratados com natalizumabe (placebo: 4,8%). Ao longo dos 2 anos de duração dos estudos, 43,5% dos pacientes tratados com natalizumabe relataram reações adversas ao medicamento (placebo: 39,6%).
As reações adversas mais frequentes que resultaram em intervenção clínica (isto é, descontinuação do tratamento com TYSABRI™) em estudos de EM foram urticaria (1%) e outras reações de hipersensibilidade (1%). Em estudos com pacientes não portadores de EM, essas reações foram exacerbação da doença de base (4,2%) e reações de hipersensibilidade aguda (1,5%).
As reações adversas de maior incidência identificadas nos estudos controlados por placebo em pacientes com EM com o natalizumabe administrado na dose recomendada são: tontura, náuseas, urticária e rigidez associadas às infusões.
As reações adversas graves mais comuns em estudos de EM com TYSABRI™ (natalizumabe) foram infecções (3,2% versus 2,6% com placebo, inclusive infecção do trato urinário [0,8% versus 0,3%] e pneumonia [0,6% versus 0%]), reações de hipersensibilidade aguda (1,1% versus 0,3%, inclusive anafilaxia [0,8% versus 0%]), depressão (1,0% versus 1,0%, incluindo ideação ou tentativas suicidas [0,6% versus 0,3%]), e colelitíase (1,0% versus 0,3%). Apendicite também foi uma reação adversa comum em pacientes que receberam TYSABRI™ (natalizumabe) (0,8% versus 0,2% com placebo).
A tabela 2 enumera as reações adversas e as anormalidades laboratoriais observadas em um estudo com pacientes de EM em uso de TYSABRI™ (natalizumabe) com uma incidência maior do que 1%, em comparação com placebo.
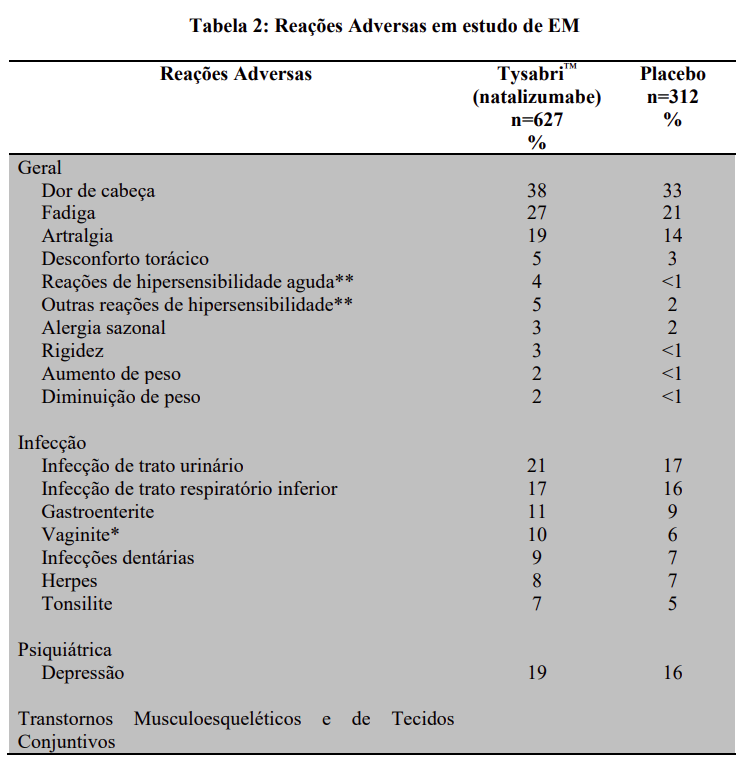
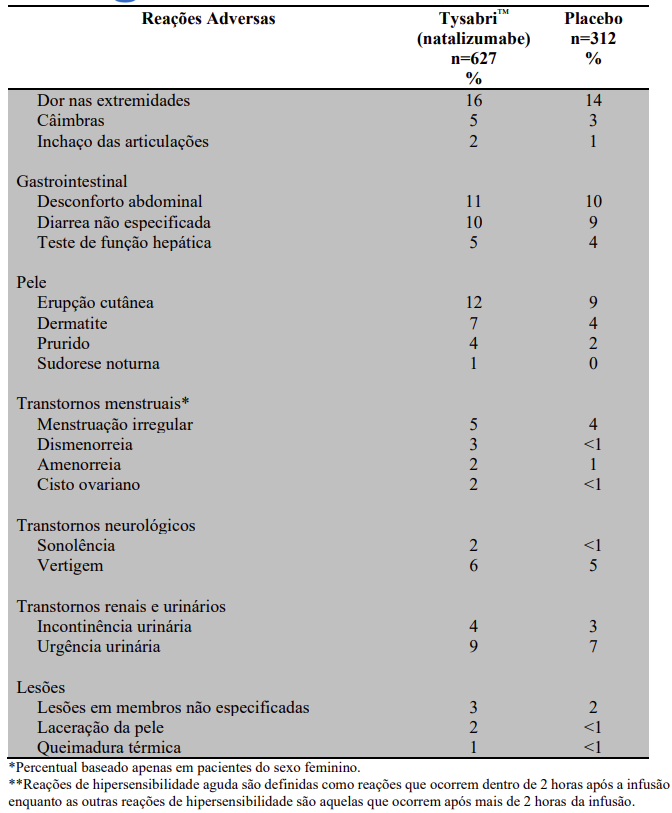
Em outro estudo com pacientes portadores de EM edema periférico ocorreu em maior frequência em pacientes que receberam TYSABRI™ (natalizumabe) (5% versus 1% com placebo).
TYSABRI™ (natalizumabe) também foi estudado para outra indicação não relacionada com EM e os achados de segurança se encontram listados nas tabelas a seguir.
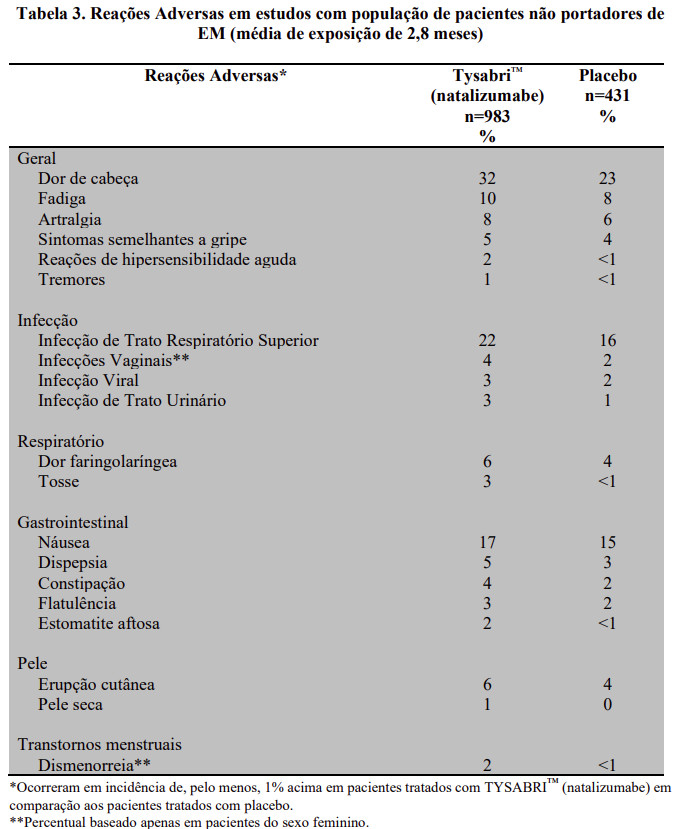

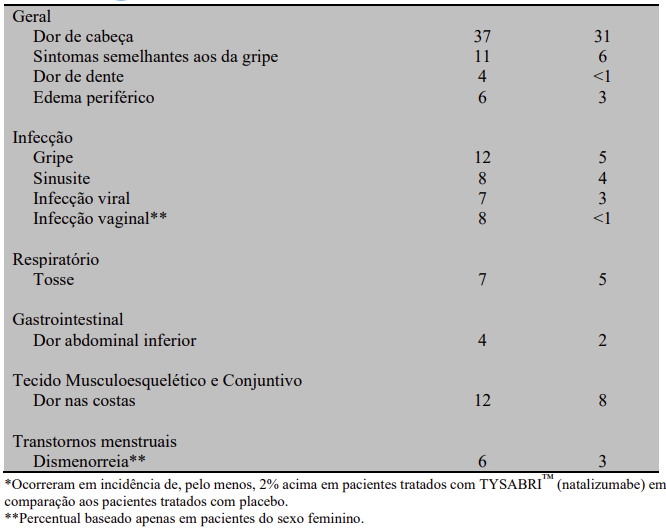
Infecções, incluindo LMP e outras infecções oportunistas
A LMP ocorreu em três pacientes que receberam TYSABRI™ (natalizumabe) em estudos clínicos. Dois casos de LMP foram observados em 1.869 pacientes portadores de EM que foram tratados, em média, por 120 semanas. Esses dois pacientes tinham recebido TYSABRI™ (natalizumabe) em adição ao tratamento com interferon beta-1a. O terceiro caso ocorreu após oito doses em um paciente dentre 1.043 pacientes não portadores de EM que foram avaliados para LMP. No cenário pós-comercialização, casos adicionais de LMP têm sido notificados em pacientes portadores de EM e não portadores de EM tratados com TYSABRI™ (natalizumabe) e que não receberam tratamento concomitante com terapia imunomoduladora.
Em estudos clínicos realizados em pacientes com EM, a taxa de infecção foi de, aproximadamente, 1,5 paciente-ano tanto em pacientes tratados com TYSABRI™ (natalizumabe) ou placebo. As infecções mais predominantes foram infecções do trato respiratório superior, gripe e infecções do trato urinário. Em um desses estudos, a incidência de infecções graves foi de, aproximadamente, 3% em pacientes tratados com TYSABRI™ (natalizumabe) comparados aos pacientes tratados com placebo. A maioria dos pacientes não interrompeu o tratamento com TYSABRI™ (natalizumabe) durante as infecções. A única infecção oportunista observada nos estudos clínicos em população de pacientes portadores de EM foi gastroenterite de curso prolongado provocada por cryptosporidium. Em estudos clínicos, a infecção por Herpes (vírus Varicella-Zoster, vírus Herpes-simplex) ocorreu numa frequência levemente maior em pacientes tratados com TYSABRI™ (natalizumabe) do que em pacientes tratados com placebo. Na experiência pós-comercialização, casos graves e às vezes fatais de encefalite e meningite causados por Herpes simplex ou Varicella-zoster foram relatados em pacientes de EM em tratamento com TYSABRI™ (natalizumabe). A duração do tratamento com TYSABRI™ (natalizumabe) antes do início dos sintomas foram de alguns meses até vários anos.
Na experiência pós-comercialização, casos raros de necrose aguda da retina (NAR) foram observados em pacientes recebendo TYSABRI™ (natalizumabe). Alguns casos ocorreram em pacientes com infecções por herpes no sistema nervoso central (por exemplo, meningite herpética e encefalite). Casos graves de NAR, afetando um ou ambos os olhos, levaram à cegueira em alguns pacientes. O tratamento relatado nestes casos incluiu terapia antiviral e, em algumas situações, cirurgia.
Em estudos conduzidos em pacientes não portadores de EM, as infecções mais comuns foram nasofaringite, infecção do trato respiratório superior e gripe. Infecções oportunistas (pneumocystis carinii pneumonia, pulmonary mycobacterium avium intracellulare, bronchopulmonary aspergillosis, e burkholderia cepacia) foram observadas em <1% dos pacientes tratados com TYSABRI™ (natalizumabe); alguns desses pacientes receberam tratamento concomitante com imunossupressores. Dois casos graves de meningites nãobacterianas ocorreram em pacientes tratados com TYSABRI™ (natalizumabe) comparados a nenhum caso em pacientes tratados com placebo.
Casos de LMP foram relatados em estudos clínicos, estudos observacionais póscomercialização e farmacovigilância pós-comercialização. A LMP geralmente leva a incapacidade severa ou morte. Casos de NCG por JCV também foram relatados durante o uso pós-comercialização do TYSABRI™ (natalizumabe). Os sintomas de NCG causada por JCV são similares ao de LMP.
Reações à infusão
Reação à infusão foi definida nos estudos clínicos como qualquer evento adverso que ocorra dentro de duas horas a partir do início da infusão. Em estudos clínicos realizados em pacientes com EM, aproximadamente 24% dos pacientes tratados com TYSABRI™ (natalizumabe) apresentaram alguma reação à infusão comparados aos 18% dos pacientes tratados com placebo. Em estudos conduzidos com população de pacientes não portadores de EM, reações à infusão ocorreram em, aproximadamente, 11% dos pacientes tratados com TYSABRI™ (natalizumabe) comparados aos 7% dos pacientes tratados com placebo. As reações mais comuns observadas em pacientes tratados com TYSABRI™ (natalizumabe) comparados aos pacientes tratados com placebo incluíram dor de cabeça, tontura, fadiga, urticaria, prurido e rigidez. Urticaria aguda foi observada em, aproximadamente, 2% dos pacientes. Outras reações de hipersensibilidade foram observadas em 1% dos pacientes em uso de TYSABRI™ (natalizumabe). Reações de hipersensibilidade sistêmicas graves ocorreram em <1% dos pacientes. Todos os pacientes se recuperaram com tratamento e/ou descontinuação da infusão.
As reações relacionadas à infusão que foram mais comuns em pacientes não portadores de EM em uso de TYSABRI™ (natalizumabe) comparados àqueles recebendo placebo, foram dor de cabeça, náusea, urticaria, prurido e rubor. Reações infusionais graves observadas em estudos clínicos para pacientes não portadores de EM ocorreram em incidência superior a 1% nos pacientes tratados com TYSABRI™ (natalizumabe).
Pacientes que apresentaram anticorpos anti-natalizumabe persistentes estiveram mais propensos a ter reações à infusão comparados àqueles que não apresentaram tais anticorpos.
Imunogenicidade
Assim como em todas as proteínas terapêuticas, há um potencial de imunogenicidade. A detecção da formação de anticorpos é altamente dependente da sensibilidade e especificidade do ensaio. Além disso, a incidência observada de anticorpos positivos (incluindo anticorpos neutralizantes) em um ensaio pode ser influenciada por vários fatores, incluindo metodologia de ensaio, manuseio de amostras, tempo de coleta de amostras, medicações concomitantes e a doença de base. Por essas razões, a comparação da incidência de anticorpos anti-natalizumabe nos estudos descritos abaixo com a incidência de anticorpos em outros estudos ou com outros produtos pode ser equivocada.
Em estudos clínicos realizados em pacientes com EM, anticorpos anti-natalizumabe foram testados a cada 12 semanas. Os ensaios utilizados não foram capazes de detectar níveis baixo a moderados dos anticorpos anti-natalizumabe. Aproximadamente 9% dos pacientes em uso de TYSABRI™ (natalizumabe) desenvolveram anticorpos detectáveis em, pelo menos, uma ocasião durante o tratamento. Cerca de 6% dos pacientes tiveram anticorpos anti-natalizumabe positivo em mais de uma ocasião. Em torno de 82% dos pacientes que apresentaram anticorpos anti-natalizumabe persistentes desenvolveram anticorpos detectáveis nas 12 primeiras semanas. Anticorpos anti-natalizumabe foram neutralizados in vitro.
A presença de anticorpos anti-natalizumabe esteve correlacionado com a redução dos níveis plasmáticos de natalizumabe. Anticorpos persistentes resultaram em redução substancial da efetividade de TYSABRI™ (natalizumabe). O risco da progressão da incapacidade e a taxa de anual de surtos foram similares em pacientes tratados com TYSABRI™ (natalizumabe) com anticorpos persistentes e em pacientes que receberam placebo.
Reações à infusão que foram mais frequentemente associadas com anticorpos antinatalizumabe persistente foram urticaria, rigidez, náusea, vômito, dor de cabeça, rubor, tontura, prurido, tremor, sensação de frio e pirexia. Demais reações adversas comuns em pacientes com anticorpos persistentes incluíram mialgia, hipertensão, dispnea, ansiedade e taquicardia.
Se, ao final de aproximadamente 6 meses de tratamento, houver suspeita de anticorpos persistentes devido à eficácia reduzida ou devido à ocorrência de reações relacionadas com a infusão, estes podem ser detectados e confirmados com um novo teste, 6 semanas após o primeiro teste positivo. Levando-se em conta que a eficácia pode ser reduzida ou que a incidência de reações de hipersensibilidade ou reações associadas à infusão pode aumentar em um paciente com anticorpos persistentes, o tratamento deve ser suspenso em pacientes que desenvolvam este tipo de anticorpos.
Em estudos clínicos com pacientes não portadores de EM, anticorpos anti-natalizumabe foram incialmente testados na semana 12 de tratamento. Em uma proporção substancial dos pacientes, esse foi o único teste realizado considerando a duração de 12 semanas dos estudos controlados por placebo. Aproximadamente 10% dos pacientes apresentaram anticorpos anti-natalizumabe, pelo menos, em uma ocasião. Anticorpos persistentes resultaram em eficácia reduzida e em aumento das reações infusionais com sintomas que incluíram urticaria, prurido, náusea, rubor e dispnea.
A imunogenicidade a longo prazo de TYSABRI™ (natalizumabe) e os efeitos dos níveis baixos e moderados dos anticorpos anti-natalizumabe são desconhecidos.
Reações de hipersensibilidade.
Em estudos clínicos controlados, com duração de 2 anos, realizados em pacientes com EM, ocorreram reações de hipersensibilidade em no máximo 4% dos pacientes. Ocorreram reações anafiláticas/anafilactoides em menos de 1% dos pacientes que receberam TYSABRI™ (natalizumabe). As reações de hipersensibilidade ocorreram, geralmente, durante a infusão ou até 1 hora após a conclusão da infusão.
Na experiência após a introdução no mercado, foram relatadas reações de hipersensibilidade que ocorreram associadas a um ou mais dos seguintes sintomas: hipotensão ou hipertensão arterial, dor no peito, desconforto no peito, dispneia e angioedema, além dos sintomas mais habituais como erupções cutâneas e urticária.
Lesão hepática
Relatos espontâneos de lesão hepática grave, aumento das enzimas hepáticas e hiperbilirrubinemia foram reportados durante o período pós-comercialização.
Anemia e anemia hemolítica
Foram relatados casos sérios e graves de anemia e anemia hemolítica em pacientes do estudo observacional pós-comercialização que estavam em tratamento com TYSABRI™ (natalizumabe).
Neoplasia
Não foram observadas diferenças nas taxas de incidência ou na natureza de neoplasias entre pacientes tratados com natalizumabe e pacientes tratados com placebo em mais de 2 anos de tratamento. No entanto, é necessária uma observação por períodos de tratamento mais prolongados antes de se poder excluir qualquer efeito do natalizumabe sobre as neoplasias.
Alterações em testes laboratoriais
Em estudos clínicos controlados, com duração de 2 anos e pacientes portadores de EM, o tratamento com TYSABRI™ (natalizumabe) foi associado ao aumento dos linfócitos, monócitos, eosinófilos, basófilos e glóbulos vermelhos nucleados na circulação. Não foram observados aumentos nos neutrófilos. O aumento dos linfócitos, monócitos, eosinófilos e basófilos relativamente à linha basal variou entre 35% e 140% para tipos individuais de células, mas a contagem média de células manteve-se dentro dos limites normais. Durante o tratamento com TYSABRI™ (natalizumabe), foram observadas pequenas reduções nas contagens de hemoglobina (decréscimo médio de 0,6 g/dl), hematócrito (decréscimo médio de 2%) e glóbulos vermelhos (decréscimo médio de 0,1 x 106 /l). Todas as alterações nas variáveis hematológicas voltaram aos valores anteriores ao tratamento, geralmente no prazo de 16 semanas da última dose do medicamento, não tendo as alterações sido associadas a sintomas clínicos. Na experiência de uso comercial, também houve relatos de eosinofilia (contagem de eosinófilos >1.500/mm3 ) sem sintomas clínicos. Em tais casos, quando o tratamento com TYSABRI™ (natalizumabe) foi descontinuado, os níveis normais de eosinófilos foram reestabelecidos. Também houve relatos de frequência incomum de trombocitopenia e púrpura trombocitopênica imunológica (PTI).
Contraindicações
Pacientes com história de hipersensibilidade ao natalizumabe, ou a qualquer outro componente da fórmula.
Pacientes que tem Leucoencefalopatia Multifocal Progressiva (LMP).
Também é contraindicado para pacientes que apresentem maior risco de manifestação de infecções oportunistas, incluindo pacientes imunocomprometidos (aqueles que estão atualmente em tratamento com medicamentos imunossupressores (IS) ou aqueles imunocomprometidos por terapias anteriores, por exemplo com mitoxantrona ou ciclofosfamida).
A combinação de natalizumabe com betainterferonas e acetato de glatirâmer é contraindicada.
Pacientes com câncer, exceto no caso de pacientes com carcinoma das células basais cutâneas.
Fonte:
Tysabri®. [Bula]. São Paulo: Biogen Brasil Produtos Farmacêuticos Ltda. Disponível em: https://consultas.anvisa.gov.br/#/bulario/q/?numeroRegistro=169930002: 14/08/2025